February 12, 2019
Last January, we observed that the new administration had yet to upend pre-existing enforcement and regulatory trends in the pharmaceutical and medical device industries. But in the past 12 months several Trump administration priorities began to coalesce. From initiatives to address the opioid epidemic, to efforts to police patient assistance programs, to attempts to depress pharmaceutical prices, 2018 saw significant governmental attention to drug and device companies.
Not all of the administration’s attention is unwanted. Notably, there have been a few signs of scaled-back executive enforcement and regulatory activity impacting drug and device companies. For example, in December, the Department of Justice (“DOJ”) moved to dismiss nearly a dozen qui tam suits brought against pharmaceutical companies under the False Claims Act (“FCA”), carrying into action recent, well-publicized policy shifts at DOJ with regard to FCA actions. In moving to dismiss those suits, DOJ observed that the relator’s theories threatened to “undermine common industry practices [that] the federal government has determined are . . . appropriate and beneficial to federal healthcare programs and their beneficiaries.”[1] Meanwhile, the U.S. Food and Drug Administration (“FDA”) clearly has recalibrated its enforcement efforts with respect to promotional activities by drug and device companies. While Congress failed to make any significant progress toward implementing long-promised legislation regarding off-label promotion, FDA finally issued two pertinent guidance documents on that issue in June.
On the other hand, although financial recoveries from actions or investigations involving drug and device makers initiated by DOJ, the Department of Health and Human Services, Office of Inspector General (“HHS OIG”), and FDA started slowly in 2018, the latter half of the year saw several large settlements. DOJ employed aggressive law enforcement tactics to combat opioid abuse, targeting a range of individuals and entities. Notably, DOJ leveraged the FCA and the Anti-Kickback Statute to investigate and bring suits against opioid manufacturers and distributors. Further, DOJ did not pull any punches with respect to drug companies’ patient assistance programs. FDA enforcement actions relating to promotional activity remained relatively consistent with past years, focusing on only the most flagrant cases of deceptive advertising. But FDA continued to crack down on issues related to current good manufacturing practices and focused, in particular, on concerns with quality and data integrity issues. We anticipate that these 2018 priorities will continue to drive DOJ and FDA activity in 2019.
In the new year, we anticipate that the new majority in the House of Representatives will amp up the pressure on pharmaceutical companies, in particular. According to the House Committee on Oversight and Reform, the Committee already has “launched one of the most wide-ranging investigations in decades into the prescription drug industry’s pricing practices,” beginning with requests for detailed information and documents regarding pricing practices from 12 drug companies.[2] But other regulatory tea leaves for 2019 are more difficult to read because of the government shutdown, which forced FDA to furlough approximately 40% of its employees at the time. Although the agency publicly announced it would continue “vital activities . . . that are critical to ensuing public health and safety,” such as screening imported products, recalls, and pursuit of civil and criminal investigations where “public health is imminently at risk,” the shutdown halted FDA’s more routine enforcement actions and regulatory activity (e.g., issuing guidance documents) during that period.[3]
We address each of these important enforcement and regulatory developments in the drug and device space below, beginning with an overview of recent government enforcement efforts and FCA jurisprudence, then moving to relevant regulatory guidance and activity regarding promotional activities, manufacturing practices, and the AKS, and concluding with notable developments on drug pricing and for device manufacturers.
As always, we would welcome the opportunity to discuss the impact of these developments with you in more detail. Additional presentations and publications on regulatory and enforcement issues impacting drug and device makers are available on our website, including industry-specific webcasts with more in-depth discussions of the FCA and practical guidance to help companies avoid or limit legal exposure.
I. DOJ ENFORCEMENT IN THE PHARMACEUTICAL AND MEDICAL DEVICE INDUSTRIES
FCA resolutions delivered the bulk of financial recoveries against drug and device companies in 2018. DOJ also brought several federal Food, Drug, and Cosmetic Act (“FDCA”) enforcement actions, but Foreign Corrupt Practices Act (“FCPA”) activity in the drug and device space remained relatively quiet this past year.
As the new year progresses, we expect to see the continued impact of two memoranda on DOJ enforcement efforts issued last January by the then-Associate Attorney General, Rachel Brand, (the so-called “Brand Memo”) and the Director of DOJ’s Civil Division’s Fraud Section, Michael Granston (the “Granston Memo”). As we detailed in our client alert, DOJ Policy Statements Signal Changes in False Claims Act Enforcement, the Brand Memo bars DOJ attorneys from using guidance documents to “create binding requirements that do not already exist by statute or regulation,” and from using DOJ’s “enforcement authority to effectively convert agency guidance documents into binding rules.”[4] But the Brand Memo acknowledges that federal prosecutors may use such guidance as evidence of scienter (e.g., as proof that a defendant knew of obligations under the law).[5]
Many agencies, including FDA (in the off-label and quality and manufacturing space) and HHS OIG (in the application of the AKS), routinely issue guidance documents interpreting relevant legislation and regulations. DOJ historically relied on these guidance documents to bolster claims that the actions of a drug or device maker violated a particular statute or regulation and thus resulted in “false” claims under the FCA. Under the Brand Memo, DOJ will have to identify other means to support its allegations, and this may well constrain enforcement activity.
The Granston Memo directs government lawyers evaluating whether to decline to intervene in a qui tam FCA action to “consider whether the government’s interests are served” by dismissal of the underlying qui tam claims pursuant to 31 U.S.C. § 3730(c)(2)(A) based on seven enumerated factors.[6] DOJ’s rollout of an updated United States Attorneys’ Manual—now entitled the Justice Manual—builds on that initiative. Indeed, the Justice Manual appears to incorporate the Granston Memo’s rubric by encouraging government lawyers to evaluate a non-exhaustive list of factors (any one of which may support dismissal) when assessing whether to intervene.[7]
Taken together, these policy statements indicate that DOJ intends to hew more closely to constitutionally defined bounds for the executive branch and to employ more judicious enforcement of the FCA—both welcome developments for drug and device manufacturers. As detailed further below, DOJ sought to dismiss a dozen qui tam suits in this past year, thereby executing on the policies embodied by the Granston Memo and the revisions to the Justice Manual.
One additional DOJ policy development merits attention. In September 2015, then-Deputy Attorney General Sally Yates issued a memorandum (the so-called “Yates Memo”) stating that corporations must provide “all relevant facts” about individuals involved in misconduct.[8] Unlike the Granston Memo, which has had a quick impact on multiple qui tam suits, the Yates Memo had a less evident impact on drug and device companies. Nevertheless, the requirement that a company identify and provide evidence on all potentially culpable individuals struck many as a significant burden. In a speech in November 2018, Deputy Attorney General Rod Rosenstein announced a fine-tuning of the Yates Memo’s requirements. Under the new DOJ policy, companies seeking cooperation credit must only identify individuals who were “substantially involved in or responsible for the criminal conduct” under investigation in white-collar investigations.[9]
A. False Claims Act
This year, DOJ recovered more than $1.1 billion from 19 FCA-related settlements with companies in the drug and device industries. Although DOJ’s civil recoveries through the first half of 2018 were less than a quarter of what they were at the same time last year, two large-scale settlements with pharmaceutical companies in the second half of the year pushed the total recoveries from resolutions with drug and device manufacturers closer to last year’s total of approximately $1.4 billion.
As illustrated below, DOJ settled 13 matters with device manufacturers (including two with AngioDynamics) and only six with pharmaceutical companies. Despite this disparity, the vast majority of DOJ’s settlement recoveries, roughly 90%, resulted from the investigations involving pharmaceutical companies. Recoveries in 2018 (like 2017) came almost exclusively from actions pursued under improper billing and AKS theories.[10]
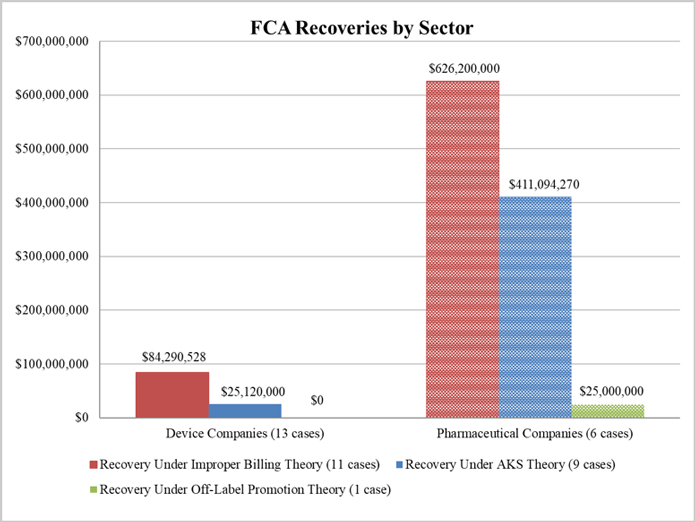
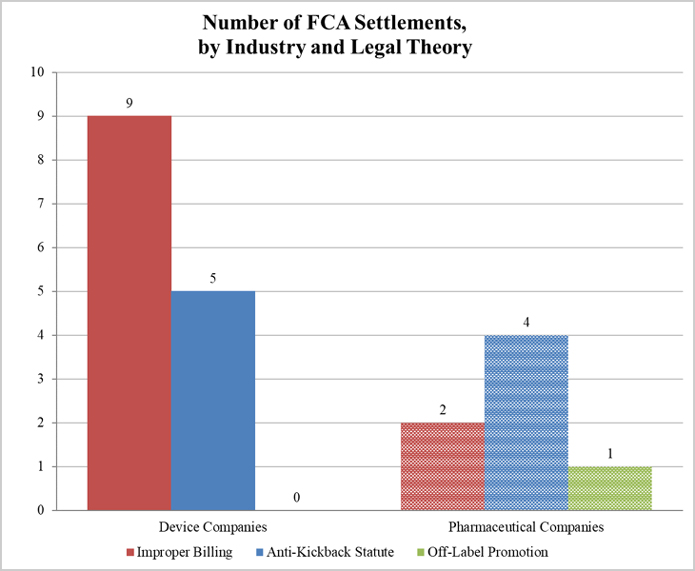
1. Settlements in FCA Matters Relating to Patient Assistance Programs
The second largest settlement in 2018 resulted from a DOJ investigation into a pharmaceutical company’s financial ties to nonprofit patient assistance programs, which help needy patients access free or low-cost medications. Further, over the past year, a second pharmaceutical company disclosed a resolution tied to a similar investigation, and two other companies disclosed resolutions in principle. These resolutions follow United Therapeutics Corp.’s $210 million settlement in December 2017, discussed in our 2017 Year-End Update.
As we reported last year, government scrutiny of patient assistance programs is a relatively recent development in the enforcement arena. Companies have supported patient assistance programs directly and indirectly for years, and the government generally has approved of such arrangements. For example, in a Special Advisory Bulletin in 2005, HHS OIG explained that certain cost-sharing assistance through “bona fide, independent charities unaffiliated with pharmaceutical manufacturers should not raise [AKS] concerns, even if the charities receive manufacturer contributions.”[11] In recent years, however, HHS OIG has determined that aspects of patient assistance programs can be problematic and raise AKS concerns.[12] Carrying on the trend we observed in our 2017 Year-End Update, several of the settlements announced this year began with investigations launched by the U.S. Attorney’s Office for the District of Massachusetts.
In December, DOJ announced its most recent resolution in this arena, which happens to be the largest to date. According to the government, Actelion Pharmaceuticals has agreed to pay $360 million to resolve claims that it used a charitable foundation to cover the copays of patients taking its pulmonary arterial hypertension drugs.[13] The government claimed that Actelion obtained data from the foundation regarding the payments the foundation made on behalf of patients taking the drugs and used that data to contribute amounts sufficient to cover the copays of only those patients, thereby purportedly violating the AKS.[14] The government further alleged that Actelion referred Medicare patients to the foundation rather than allowing them to participate in the company’s free drug program in order to generate revenue.[15]
The first half of the year saw several other similar matters reach the resolution stage:
- In May, Pfizer, Inc. announced it had agreed to pay $23.85 million to resolve allegations that it violated the AKS by paying patients’ copay obligations with funds purportedly channeled through a foundation.[16] According to the government, Pfizer donated money to the foundation for the purpose of covering copays for two of its drugs used to treat renal cell carcinoma, which could have been provided at no cost to patients who qualified for Pfizer’s existing free drug program.[17] The government claimed that paying patients’ copay obligations constituted remuneration designed to induce patients to use Pfizer’s drugs.[18] Further, according to the government, Pfizer worked with the foundation to create and finance a fund for patients suffering from the heart condition treated by its drug Tikosyn.[19] Pfizer allegedly referred patients to the fund and timed the creation of the fund to coincide with Pfizer’s increase in the price of the drug.[20] The government asserted that these actions allowed Pfizer to generate more revenue, while masking the effect of its price increases.[21]As part of the resolution, Pfizer entered into a five-year corporate integrity agreement (“CIA”) with HHS OIG, in which Pfizer agreed to ensure legally compliant interactions with third-party patient assistance programs.[22] Intended to “promote[] independence” between the company and any such programs to which it donates, the agreement requires review by an independent organization, compliance-rated certifications, and the implementation of a risk assessment and mitigation process.[23]
- Also in May, Dublin-based Jazz Pharmaceuticals PLC announced in an SEC filing that it had agreed in principle to pay $57 million to resolve similar allegations.[24] Neither Jazz nor the government has disclosed the government’s allegations in public sources, but the company’s SEC filings indicate that the allegations generally concern Jazz’s financial support of patient assistance nonprofits that help cover copays.[25] The company had not finalized the settlement with DOJ at the time of its most recent quarterly filing.[26]
- In early June, Denmark-based H. Lundbeck A/S announced that its U.S. subsidiary, Lundbeck LLC, had reached an agreement in principle with DOJ to pay $52.6 million to resolve DOJ’s investigation into the company’s ties to patient assistance charities.[27] Although the final terms of the agreement remain subject to negotiation and few details are publicly available,[28] Lundbeck previously disclosed that its subsidiary, Lundbeck NA Ltd., received a subpoena from the U.S. Attorney’s Office for the District of Massachusetts in 2016, relating to its “payments to charitable organizations providing financial assistance to patients taking Lundbeck products” and to sales and marketing practices.[29]
These patient assistance investigations raise a host of legal and policy questions, ranging from the appropriate reach of the AKS to the impact on bona fide corporate charitable contributions to the First Amendment implications of the government’s theories. In light of these key questions, it came as no surprise that a charity, Patient Services Inc., filed a First Amendment lawsuit in the U.S. District Court for the Eastern District of Virginia in January challenging the constitutionality of HHS OIG’s restrictions on the charity’s communications with donors.[30] Despite the thorny legal and policy questions, it appears DOJ will continue pursuing claims tied to patient assistance programs.
2. Settlements in AKS-Related FCA Matters
In addition to its activity relating to patient assistance programs, DOJ reached a number of other notable AKS-related settlements in 2018.
In August, Insys Therapeutics reached an agreement in principle with the government to pay at least $150 million, and potentially an additional $75 million depending on certain events, to resolve a DOJ investigation into claims that the drug maker paid kickbacks to doctors to induce them to prescribe Subsys, an opioid medication.[31] According to the government’s complaint in intervention, Insys allegedly paid doctors sham speaker fees, hired doctors’ friends and relatives, and provided doctors with lavish meals and entertainment to encourage more Subsys prescriptions.[32] The government further asserted that Insys caused federal health care programs to pay for uses of Subsys that are not covered by the programs; for example, the government contended that Insys encouraged doctors to prescribe Subsys even when it was not medically necessary and also misrepresented patients’ medical diagnoses to federal program sponsors.[33] A number of former Insys employees and doctors previously pled guilty to criminal charges relating to alleged kickbacks for Subsys prescriptions,[34] and the company’s former Vice President of Sales pled guilty to one count of racketeering conspiracy in November 2018.[35] The company’s former CEO also pled guilty to conspiracy and mail fraud charges in January 2019,[36] and the trial of several other former executives is currently underway.
The government announced in October that Abbott Laboratories and AbbVie Inc. agreed to pay $25 million to settle claims that Abbott sales representatives provided doctors with gift baskets, gift cards, and other items in exchange for prescriptions of the drug TriCor.[37] The settlement also resolved claims that Abbott marketed and promoted TriCor for unapproved uses, including treatment of diabetic patients and the reduction of cardiac health risks.[38]
In December, the government announced that Covidien (which Medtronic acquired in 2015), agreed to pay $13 million to resolve claims under the FCA stemming from efforts to collect data regarding user experiences with its device.[39] Additionally, Medtronic will pay another $20 million to resolve other DOJ claims “concerning various market-development and physician engagement activities” conducted by Covidien and ev3, which Covidien acquired in 2010.[40]
In addition to the actions above, DOJ announced settlements of several other AKS-related cases with drug and device makers throughout the year:
- In March, Abiomed, Inc. agreed to a $3.1 million settlement to resolve claims that it paid for expensive meals to induce physicians to use the company’s line of heart pumps.[41] The government claimed that Abiomed managers approved expenses for meals even though the cost-per-attendee far exceeded internal company expense guidelines, the company purportedly misrepresented the number of attendees and/or listed fictitious names to make costs appear lower, and attendees ordered alcohol in amounts inconsistent with legitimate scientific discussion.[42] This resolution continues a line of matters in which DOJ has pointed to meal expenses that surpassed company limits as evidence of improper remuneration.[43]
- In May, the government intervened to take part in a $1.9 million settlement with medical equipment supplier Precision Medical Products, Inc. (“PMP”).[44] Between January 2011 and 2017, PMP allegedly based its independent contractor sales representatives’ commissions on the amount of federal health program reimbursements they could obtain.[45] (Notably, HHS OIG rejected expanding the bona fide employee safe harbor to this contract sales force.) The complaint alleged that these acts were illegal kickbacks because they incentivized referrals of patients and patient orders to PMP that may be covered by government-funded programs.[46] PMP also allegedly forged documents to obtain approvals and the related reimbursements for its durable medical equipment (“DME”) products in violation of federal program requirements,[47] including through techniques dubbed “magic time” (i.e., tracing a physician’s signature onto other documents) and “ninja drive” (i.e., using a presigned form).[48] The complaint further asserts that PMP violated federal rules by waiving co-insurance payments and then billing the government.[49]
- In August, Trinity Medical Pharmacy and several of its principals agreed to pay more than $2.24 million to resolve allegations that it knowingly billed government programs for claims driven by kickbacks and omitted its COO’s previous felony conviction from its application to become a certified provider with Express Scripts, the pharmacy benefit manager for TRICARE and several other carriers.[50]
- Also in August, Florida-based provider of oxygen and respiratory therapy services Lincare paid $5.25 million to settle claims that it improperly waived or reduced co-insurance, co-payments, and deductibles for beneficiaries of a Medicare Advantage Plan and caused the submission of false claims for payments to Medicare.[51]
- In December, medical device company LivaNova USA agreed to pay $1.87 million to resolve allegations that it knowingly paid kickbacks in the form of speaking fees to Georgia physicians to induce them to refer its devices for treatment of refractory epilepsy.[52]
3. Other Settlements in FCA Matters
The remainder of the government’s settlement recoveries during 2018 came from alleged violations of government health program requirements or restrictions on off-label promotion.
In October, the government announced the largest FCA enforcement settlement of the year, with AmerisourceBergen Corporation and four of its subsidiaries (“ABC”).[53] ABC agreed to pay $625 million to resolve allegations that a facility it operated repackaged oncology-supportive drugs into pre-filled syringes and distributed those syringes to doctors to provide to cancer patients.[54] The government alleged that ABC sought to profit from the operation by purchasing the syringes from manufacturers, pooling their contents, repackaging the drugs, and harvesting the overfill to create more doses than it originally purchased.[55] The government further claimed that ABC was able to bill multiple health care providers for the same syringe and increase its market share by offering discounts.[56] This settlement follows a 2017 settlement in which one of ABC’s subsidiaries pled guilty to illegally distributing misbranded drugs and agreed to pay $260 million to resolve criminal claims that it distributed the drugs from a facility that was not registered with FDA.[57]
Although the ABC resolution dwarfs DOJ’s other settlements in this space, the past year saw multiple additional enforcement actions involving purported violations of federal program requirements and/or off-label promotion:
- In March, Alere Inc. and its subsidiary, Alere San Diego, agreed to pay $33.2 million to resolve allegations related to sales of purportedly unreliable diagnostic devices.[58] According to the government, Alere received customer complaints that put it on notice that certain of its devices used to diagnose serious heart conditions potentially transmitted false positives and false negatives; nevertheless, the government alleged, the company did not take appropriate corrective actions until government inspections resulted in a nationwide recall.[59] Of the settlement, more than $4.8 million will be returned to individual states.[60]
- In April, Allergan, Inc. settled claims involving allegations that the company sold purportedly defective weight loss devices, marketed the device for use in surgical procedures that were not described in the product’s approved labeling, and made inappropriate payment to physicians.[61] Allergan agreed to pay $3.5 million in connection with the resolution, nearly $200,000 of which will be split among impacted states.[62]
- In July, medical device manufacturer AngioDynamics agreed to pay $11.5 million to resolve allegations that it marketed a product for unapproved uses and instructed health care providers to use incorrect billing codes to submit claims for those uses.[63] It also agreed to pay an additional $1 million to settle claims that certain AngioDynamics employees represented to health care providers that federal health care programs would cover an unapproved use of a recalled and re-issued product previously used to treat perforator veins, thus causing false claims to be submitted to the programs.[64]
- In September, the government announced that now-defunct compounding pharmacy RS Compounding and its owner agreed to pay $1.2 million to resolve allegations that it charged federal health care programs a higher price for its drugs (in some cases, over 10,000 percent higher) than it charged the public.[65]
- In October, Cooley Medical Equipment agreed to pay more than $5.25 million to settle claims that it falsely stated that certain of its compounded medical creams used cream-based ingredients rather than bulk powder, to avoid prior authorization requirements for bulk powder ingredients or limited reimbursement from federal health care programs.[66] Cooley self-disclosed the conduct and will be allowed to pay the settlement amount—which, according to the government, is 1.5 times the amount of monetary loss caused by the false claims—over a period of six years.[67] Because the government typically insists on doubling purported damages in settlement discussions, it appears that the government underscored the loss calculation so as to encourage other entities to self-disclose and cooperate.
B. Developments in Enforcement Actions Against Opioid Manufacturers and Distributors
As reported in our 2017 Mid-Year and Year-End updates, DOJ has made criminal and civil enforcement related to the opioid epidemic a top priority. Reflecting on developments in 2018, it is clear that DOJ is following through on its commitment to target aggressively the opioid epidemic, and all signs indicate that these efforts are likely to persist for the foreseeable future.
Numerous comments by DOJ officials suggest that DOJ is prepared to pursue litigation against manufacturers in conjunction with the Department’s efforts to stem the tide of opioid misuse, overdoses, and deaths. On February 27, 2018, DOJ announced the creation of the Prescription Interdiction & Litigation (“PIL”) Task Force, which “will aggressively deploy and coordinate all available criminal and civil law enforcement tools” to curb the opioid epidemic, “with a particular focus on opioid manufacturers and distributors.”[68] In remarks delivered the following day, Deputy Assistant Attorney General Stephen Cox specifically stated that DOJ would be using the FCA as a tool to address the opioid epidemic.[69] Just last month, Deputy Assistant General James Burnham also commented that the FCA and FDCA “are squarely implicated” should a pharmaceutical company misrepresent opioid-related risks; he cited potential enforcement avenues against pharmaceutical companies, including actions under the Controlled Substances Act (“CSA”) for failure to report suspicious orders of controlled substances and misbranding-related actions under the FDCA for failure to communicate adequate risk information in accordance with FDA-required Risk Evaluation and Mitigation Strategies.[70]
DOJ also announced a program this year to target suppliers and wholesale distribution networks responsible for distributing fentanyl and other synthetic opioids, which will focus on ten districts with high rates of deadly drug overdoses.[71] In support of these enforcement efforts, DOJ is utilizing a new data analytics program focusing specifically on opioid-related health care fraud, which enables DOJ officials to pinpoint areas with high rates of prescription, dispensing, and overdoses.[72]
In some cases, DOJ’s heated rhetoric has previewed hard-hitting acts. In April, DOJ intervened in now-consolidated suits alleging AKS-related violations against Insys Therapeutics, Inc., the manufacturer of a sublingual fentanyl spray approved for the treatment of breakthrough pain in adult cancer patients already on opioid therapy. In addition to the AKS-related claims noted above, the complaint alleged that the manufacturer encouraged off-label use of the product in situations where the use was “not medically reasonable and necessary” and misrepresented patient diagnoses to secure reimbursement for the company’s product.[73] As discussed above, Insys Therapeutics reached an agreement in principle with the government to pay at least $150 million and potentially up to $225 million to resolve the claims.[74] Several former Insys employees and doctors previously pled guilty to criminal charges relating to alleged kickbacks for Subsys prescriptions,[75] the company’s former Vice President of Sales pled guilty to one count of racketeering conspiracy in November 2018,[76] and the company’s former CEO recently pled guilty to conspiracy and mail fraud charges in January 2019.[77]
Further underscoring the government’s ongoing scrutiny of the industry, other manufacturers also have disclosed that they are in receipt of subpoenas related to the manufacture, distribution, and marketing of opioids, including three manufacturers that received subpoenas from the U.S. Attorney’s Office for the Southern District of Florida related to generic opioid products.[78]
In April, DOJ also filed a “friend of the court” motion in ongoing multidistrict litigation against opioid manufacturers and distributors to enable it to participate in settlement negotiations.[79] Before filing its motion, DOJ announced that it would seek reimbursement from the defendants for the costs that the federal government has incurred as a result of the opioid epidemic, such as costs borne by federal health care programs and the expenses associated with law enforcement initiatives.[80] And, in August, DOJ announced the unsealing of an indictment charging two Chinese citizens with operating a conspiracy through numerous companies (including companies within the United States) to manufacture and ship fentanyl analogues and other drugs illegally to the United States.[81]
Of note, DOJ has taken steps to address the opioid epidemic from a regulatory perspective as well. Observing that the United States is an outlier in the number of opioids prescribed each year, former Attorney General Jeff Sessions issued a memo directing the U.S. Drug Enforcement Administration (“DEA”) to evaluate whether to amend regulations addressing the aggregate production quota, which sets the quantity of opioids that manufacturers are permitted to produce each year.[82] In response, the DEA issued a proposed rule that would limit opioid production in some instances and strengthen controls to prevent diversion of controlled substances.[83] Later in the year, in an effort to “encourage vigilance on the part of opioid manufacturers,” DOJ and the DEA proposed an average ten percent reduction in 2019 manufacturing quotas for the six most frequently misused opioids.[84]
On October 24, 2018, President Trump also signed into law The Substance Use – Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities Act (the “SUPPORT for Patients and Communities Act”), which seeks to “reduc[e] access to the supply of opioids by expanding access to prevention, treatment, and recovery services.”[85] Among other items, the bipartisan legislation bolsters requirements for CSA registrants to design and operate systems to identify suspicious orders and report those orders to the DEA,[86] requires drug manufacturers and distributors to review quarterly DEA reports showing the total number of registrants distributing controlled substances as well as the total quantity and type of opioids distributed to pharmacies and practitioners,[87] requires HHS to support state efforts to develop their own prescription drug monitoring programs, and allows FDA to recall a controlled substance if it determines there is a “reasonable probability” that the controlled substance “would cause serious adverse health consequences or death.”[88]
C. Notable Developments in FDCA Enforcement
In 2018, DOJ obtained a number of injunctions against and resolutions with manufacturers and distributors to prevent the sale and distribution of allegedly unapproved, adulterated, and misbranded drugs or devices.
In December, DOJ secured guilty pleas—and a significant fine—in an FDCA case alleging that Tokyo-based Olympus Medical Systems Corporation and a former senior executive distributed misbranded medical devices in interstate commerce.[89] According to the government (and the plea documents), Olympus failed to file adverse event reports, known as Medical Device Reports (“MDRs”), with FDA in 2012 and 2013 after learning that certain patients using its duodenscopes contracted infections.[90] Olympus allegedly also received an independent expert report in 2012 that identified the possibility of bacteria in the duodenscopes and recommended further investigation and updated cleaning instructions.[91] Under the FDCA, devices with required but unfiled MDRs or supplemental MDRs are deemed misbranded.[92] Pursuant to the plea deal, the U.S. District Court for the District of New Jersey fined Olympus $80 million and ordered $5 million in criminal forfeiture. Olympus also agreed to implement a number of compliance measures, including the retention of an MDR expert who will review Olympus’s compliance with the FDCA’s MDR requirements, as well as periodic review by the company’s president and board of directors.[93] The sentencing of the former executive is set for March 2019.[94]
DOJ also pursued several drug and device makers for alleged promotion of unsupported therapeutic claims and other unapproved uses.
In March, for example, DOJ announced that the U.S. District Court for the Middle District of Florida permanently enjoined a Florida company, MyNicNaxs LLC, and two associated individuals from selling sexual enhancement and weight loss products until the company institutes specific remedial measures and obtains written approval from FDA.[95] According to the complaint, the products contained undisclosed ingredients, some of which have been shown to increase the risk of heart attacks and strokes. Further, the government alleged that no scientific evidence supported the company’s claims that the products cured or prevented serious diseases.[96]
The U.S. District Court for the District of New Jersey in August similarly enjoined two companies—S Hackett Marketing LLC d/b/a Just Enhance of Trenton, New Jersey and R Thomas Marketing LLC of Bronx, New York—and related individuals from selling unapproved sexual enhancement products that the government claimed contained an undisclosed ingredient and also lacked labels revealing potentially adverse consequences of using the products.[97]
In July, three related Chicago companies, their owner, and their operations manager also agreed to settle claims that they were manufacturing, selling, and distributing adulterated and misbranded dietary supplements and unapproved drugs and to be bound by a consent decree of permanent injunction.[98] The government’s complaint alleged that the companies’ claims that their products could help prevent or treat diseases such as Alzheimer’s, diabetes, HIV, and Parkinson’s were unsupported by credible scientific evidence, labels were deficient, and FDA inspections revealed failures to comply with current good manufacturing practice regulations, including failure to establish written sanitation procedures.[99]
In early December, Minnesota-based medical device manufacturer ev3 Inc. agreed to plead guilty to a misdemeanor charge in connection with distribution of its liquid embolization device, Onyx.[100] The government alleged that, although FDA approved Onyx only for use inside the brain, ev3 sales representatives encouraged surgeons to use Onyx for other, unapproved uses from 2005 to 2009.[101] The government further claimed that ev3’s management designed sales quotas and bonuses that incentivized sales of Onyx for such uses.[102] As part of the resolution, ev3 will pay a fine of $11.9 million, and it will forfeit $6 million.[103] Medtronic, ev3’s current parent company, also has agreed to implement new compensation structures and conduct compliance monitoring relating to Onyx sales and marketing.[104] As noted above, DOJ separately entered into a civil settlement with Covidien related to allegations brought under the FCA.
DOJ also took action against several companies in connection with purported current good manufacturing practice (“cGMP”) violations:
- As described in more detail below, DOJ announced consent decrees permanently preventing distribution of adulterated drugs by two compounding pharmacies, Cantrell Drug Company and Delta Pharma, Inc., and related individuals.[105] DOJ alleged that the drugs were adulterated because the companies failed to comply with cGMP requirements and because the drugs may have been contaminated as a result of insanitary conditions.
- In October, DOJ also announced consent decrees of permanent injunctions to prevent distribution of misbranded drugs against Keystone Laboratories, Inc. and its owner and operator.[106] DOJ alleged that Keystone distributed hair and skin care products that failed to comply with FDA’s cGMPs due to potential bacterial contamination from insanitary conditions.[107] The company cannot begin manufacturing again unless it complies with specific remedial measures, and the injunction provides for safeguards if the defendants work with third parties to manufacture their products.[108]
D. FCPA Investigations
Although multiple DOJ and SEC investigations into the foreign sales practices of many drug and device companies remain ongoing, DOJ did not announce any FCPA settlements with drug and device companies in 2018. This quiet may not last long: DOJ previously signaled that it plans to ramp up its FCPA enforcement in the health care space, as discussed in our 2017 Year-End Update. In a speech delivered in 2017, Sandra Moser, then-Acting Chief of DOJ’s Fraud Section, announced that prosecutors from DOJ’s FCPA unit would begin “working hand in hand” with the Corporate Strike Force of DOJ’s Healthcare Fraud Unit to “investigate and prosecute matters relating to healthcare bribery schemes, both domestic and abroad.”[109]
For its part, the SEC announced two FCPA settlements with drug and device companies in 2018.
- On September 4, a Paris-based pharmaceutical company agreed to pay more than $25 million in disgorgement, prejudgment interest, and penalties to resolve FCPA-related charges by the SEC regarding alleged corrupt payments to government procurement officials and health care providers in Kazakhstan and the Middle East. The company also agreed as part of the resolution to self-report about anti-corruption compliance to the SEC for a two-year period.[110]
- On September 28, a Michigan-based medical device company settled FCPA-related charges by the SEC. According to the SEC, the company violated the books and records and internal accounting controls provisions of the FCPA. The company agreed to pay a $7.8 million penalty, and the settlement was the SEC’s second FCPA action against the company. The company did not admit or deny the SEC’s charges, but consented to a cease-and-desist order and the penalty.[111]
II. FCA JURISPRUDENCE DEVELOPMENTS RELEVANT TO DRUG AND DEVICE COMPANIES
A. Developments in the Implied Certification Theory’s Materiality Requirement
As detailed in our 2017 Mid-Year and Year-End False Claims Act Updates, the federal courts continue to grapple with the implications of the Supreme Court’s decision in Universal Health Servs., Inc. v. United States ex rel. Escobar (“Escobar”).
Under the theory of falsity addressed in Escobar, a company impliedly certifies compliance with all conditions of payment when it submits a claim to the government and, if that claim fails to disclose the violation of a material statutory, regulatory, or contractual requirement, the omission renders the claim false in violation of the FCA. In Escobar, the Supreme Court held that the implied false certification theory can be a basis for FCA liability if two conditions are met:
- “[F]irst, the claim does not merely request payment, but also makes specific representations about the goods or services provided; and
- [S]econd, the defendant’s failure to disclose noncompliance with material statutory, regulatory, or contractual requirements makes those representations misleading half-truths.”[112]
One area of focus in post-Escobar cases involving companies in the health care industry has been the question of what, if any, impact the government’s continued payment of claims should have on the materiality analysis.
In June, for instance, the U.S. District Court for the Northern District of Illinois allowed an FCA claim by the government to proceed against Snap Diagnostics, the manufacturer of a home sleep apnea test.[113] The government alleged that the company encouraged unnecessary tests to be billed to Medicare where only a single test was required. According to the government, the company, in lobbying the government for Medicare coverage of its testing, advised that it was routine to conduct only one night of sleep apnea testing, and the company’s promotional materials stated that a diagnosis can almost always be made in a single night of testing. But the company’s alleged procedure was to bill for multiple nights of testing only when the patient was Medicare-covered or self-pay, but not when the patient had private insurance. Further, the government asserted that the decision was not based on a clinical determination of medical necessity. Additionally, the government pointed to Medicare guidance stating that Medicare will cover a second or third night of sleep apnea testing only when it is medically necessary.
Addressing materiality at the motion-to-dismiss stage, the court rejected the argument that the government’s routine payment of multiple claims served as evidence that the requirements were not material. The court not only concluded that this argument was premature at the pleading phase, but also described this argument as a “logical falsity” that misstates the Supreme Court’s position in Escobar.[114] Relying on the Seventh Circuit’s decision in United States v. Sanford-Brown, Ltd.,[115] the court ruled that the government met its burden in showing materiality. Because Medicare guidance provided for reimbursement of a second or third night of sleep apnea testing only when medically necessary, the court found that the company’s submission of claims for subsequent nights of testing could constitute a misleading representation that those nights had been determined to be medically necessary and permitted the claim to go forward.
In another matter implicating the materiality analysis post-Escobar, the Supreme Court recently denied certiorari in a case in which Gilead Sciences, Inc. sought review of a Ninth Circuit decision allowing a qui tam suit to continue past the motion to dismiss stage (United States ex rel. Campie v. Gilead Sciences, Inc.).[116] The Ninth Circuit held that the relators adequately alleged that Gilead fraudulently obtained FDA approval for certain of its drugs by making purportedly false statements to FDA about the source of the drugs’ active ingredient and the ingredient’s compliance with FDA regulatory requirements. Because Medicare and Medicaid reimbursement for drugs is contingent upon FDA approval, which Gilead allegedly obtained fraudulently, the Ninth Circuit concluded that Gilead’s submission of claims for payment for “FDA approved” drugs amounted to a material misrepresentation that could serve as a basis for liability under the FCA. The Ninth Circuit rejected Gilead’s argument that, because FDA did not rescind approval of the drug after learning of the violations and the Centers for Medicare and Medicaid Services continued to pay for the drug, the violations were not material to the government’s payment decision. Instead, the court reasoned that Gilead ultimately stopped using the active ingredient from the unapproved manufacturer, and “[o]nce the unapproved and contaminated drugs were no longer being used, the government’s decision to keep paying for compliant drugs does not have the same significance as if the government continued to pay despite continued noncompliance.”[117]
Gilead’s petition for certiorari presented the question of whether the government’s decision to continue reimbursement after learning of alleged regulatory infractions should presumptively defeat a relator’s claim that the regulatory infractions were material to the government’s payment decision.[118] Gilead maintained that the Ninth Circuit’s treatment of this issue clashes with the Supreme Court’s instruction in Escobar that “if the Government pays a particular claim in full despite its actual knowledge that certain requirements were violated, that is very strong evidence that those requirements are not material.”[119]
As discussed in our prior updates, the Supreme Court invited the Solicitor General to present the government’s position on the case, thereby signaling interest in the impact of government acquiescence on efforts by the government or relators to establish materiality. In response, the Solicitor General argued strongly that the Court should not grant certiorari and indicated that it would move to dismiss the case if it were sent back to the district court.[120] Arguing that the case is “not in the public interest,” DOJ explained that its position was based on a “thorough investigation” of the merits of the case, as well as concerns about the “burdensome” discovery requests that the parties would direct at FDA if the case were to move forward.[121] With the Supreme Court’s denial of certiorari, the case has returned to the district court in accordance with the Ninth Circuit’s previous decision, and DOJ has not yet moved to dismiss the case at the district court level.
Whether or not DOJ’s seemingly abrupt move before the Supreme Court reveals any deeper concerns within DOJ around potentially adverse post-Escobar rulings, it is a marked example of DOJ’s purported commitment under the Justice Manual and Granston Memo to dismissing at least some of the unmeritorious cases brought forth by qui tam relators. In the meantime, the question of government acquiescence in the context of materiality remains open for courts’ consideration.
B. Scienter under Escobar
The Supreme Court in Escobar expressed confidence that robust enforcement of the FCA’s scienter and materiality elements would prevent a broad view of falsity from subsuming the purposes of the Act. One (albeit unpublished) appellate decision this past year provides at least some support for this perspective.
In United States ex rel. Streck v. Allergan, Inc., the Third Circuit held that an FCA relator fails to plead scienter where the defendant acted based on a reasonable, albeit incorrect, interpretation of relevant statutory and regulatory guidance.[122] The relator in Streck based his FCA claims on allegations that the defendant pharmaceutical companies failed to account for “price-appreciation credits” in submitting Average Manufacturer Prices (“AMPs”) for certain drugs when calculating rebates owed by those companies to Medicaid under the Medicaid Drug Rebate Program.[123] In assessing the relator’s claims, the court first considered “whether the relevant statute was ambiguous,” then evaluated “whether [the] defendant’s interpretation of that ambiguity was objectively reasonable,” and, finally, assessed “whether [the] defendant was ‘warned away’ from that interpretation by available administrative and judicial guidance.”[124] The Third Circuit observed that the statutory definition of “price paid to the manufacturer” for AMP purposes was unclear because it did not specify whether it was the “initial” price (which would have excluded subsequent price-appreciation credits) or “cumulative” price (which would have included them).[125] Even though the defendants’ interpretation that the statute and the associated guidance excluded price-appreciation credits may not have been “the best interpretation,” the Third Circuit nonetheless concluded that the interpretation was reasonable, that there was no guidance “warn[ing] away” from that interpretation, and that holding defendants liable for a “reasonable interpretation of an ambiguous statute was inconsistent with the reckless disregard [relator] was required to allege at this stage of the litigation.”[126]
C. Rule 9(b) Particularity
Federal Rule of Civil Procedure 9(b) requires FCA plaintiffs to plead allegations of fraud with particularity. In cases brought against companies, such as drug and device manufacturers, that do not typically submit claims to government payors directly, Rule 9(b) requires an FCA complaint to adequately plead facts establishing a link between the defendant’s challenged conduct and claims for reimbursement submitted by third-party physicians or pharmacies to the government. As discussed in our 2017 Mid-Year and Year-End updates, courts have diverged over the question of what allegations suffice to survive a Rule 9(b) challenge on a motion to dismiss.
In United States ex rel. Solis v. Millennium Pharmaceuticals, Inc., the Ninth Circuit added to the body of precedent holding that FCA claims that fail to allege particularized facts linking the alleged scheme to at least one specific claim submitted to the government cannot survive a Rule 9(b) motion to dismiss.[127] The relator, a pharmaceutical sales representative, brought a qui tam FCA action against his former employer alleging, among other things, that the company paid kickbacks to physicians to prescribe the antibiotic drug Avelox. The district court dismissed all of the relator’s claims, holding that they were foreclosed by the public disclosure bar.
On appeal, the Ninth Circuit held that the district court had erred in holding that the public disclosure bar foreclosed the relator’s claim relating to Avelox because none of the public disclosures at issue mentioned this claim. Noting that it could affirm on any ground supported by the record, the Ninth Circuit nevertheless affirmed the district court’s dismissal of this claim, holding that it failed to satisfy Rule 9(b) particularity requirements. The relator’s “only particularized allegations” showed efforts by the defendant “to get Avelox placed ‘on formulary’ at two hospitals,” which “merely means the drug is available to be used or prescribed.”[128] “Even assuming these efforts established a scheme to submit false claims,” the relator neither “identif[ied] a single claim submitted pursuant to the scheme” nor provided “reliable indicia supporting a strong inference that such claims were submitted.”[129] Indeed, the Ninth Circuit noted that the relator did not even “allege that being ‘on formulary’ meant such claims would be submitted,” or that “being on formulary meant Avelox would be prescribed.”[130] Because the complaint did not contain “other details linking the alleged scheme to any claim submitted to a federal healthcare program,” the relator failed to “plead[] with the particularity required by Rule 9(b).”[131]
The Eleventh Circuit reached a similar conclusion in Carrel v. AIDS Healthcare Foundation, Inc.[132] There, the relators alleged that the foundation violated the AKS and thus the FCA by paying bonuses to its employees for referring patients with HIV/AIDS to other services provided by the foundation and for which the foundation sought federal reimbursement. In addition to rejecting the relators’ AKS theory (as described below), the Eleventh Circuit also rebuffed relators’ assertion that the district court erred in dismissing a host of relators’ claims for lack of particularity.
Long-standing Eleventh Circuit requires relators to plead the “actual ‘submission of a [false] claim’” with some “indicia of reliability”; assertions that a “claims requesting illegal payment must have been submitted, were likely submitted[,] or should have been submitted” do not suffice.[133] Applying that precedent in Carrel, the Eleventh Circuit observed that although relators “allege[d] a mosaic of circumstances that are perhaps consistent with their accusations that the Foundation made false claims . . . [they] fail[ed] to allege with particularity that these background factors ever converged and produced an actual false claim.”[134] Notably, the court batted aside relators’ efforts to rely on “mathematical probability” and their insider knowledge of the foundation’s operations, explaining that relators’ “access to possibly relevant information” did not “translate[] to knowledge of actual tainted claims presented to the government.”[135]
D. Retaliation
The FCA provides remedies to employees if they are “discharged, demoted, suspended, threatened, harassed, or in any other manner discriminated against in the terms and conditions of employment because of lawful acts” conducted in furtherance of an FCA claim.[136]
In DiFiore v. CSL Behring, LLC, the Third Circuit recently addressed the standard for showing causation in an FCA retaliation claim brought against a biopharmaceutical company. The plaintiff appealed the district court’s jury instruction requiring the plaintiff to show “protected activity was the ‘but-for’ cause of an adverse action.”[137] On appeal, the plaintiff argued that the FCA only requires proof that the protected activity was a “motivating factor” in the adverse action.
In rejecting this argument and affirming the district court, the Third Circuit relied on the Supreme Court’s analysis in Gross v. FBL Financial Services, Inc.[138] and University of Texas Southwestern Medical Center v. Nassar,[139] both of which concerned the causation standard in the discrimination context. The court observed that the statutory text “because of” was identical in the FCA to the language used in the Age Discrimination in Employment Act and Title VII’s anti-retaliation provision, and in those contexts the Supreme Court held that this language requires proof that the protected status or activity was the “but-for causation” for the employer’s adverse employment action. The court contrasted this “because of” language with the text of the anti-retaliation regulation under the Family Medical Leave Act (“FMLA”), which required the protected activity to be a “negative factor” in an employment decision. Because the FCA included the same “because of” language at issue in Gross and Nassar, rather than the “negative factor” language used in the FMLA regulation, the court held that the “but-for” standard adopted in Gross and Nassar governed its interpretation of causation under the FCA.[140]
E. Public Disclosure Bar
As amended by the Affordable Care Act in 2010, the FCA’s public disclosure bar provides that a court “shall dismiss” an FCA action if “substantially the same allegations or transactions . . . were publicly disclosed” through listed sources, as long as the relator does not qualify as an “original source.”[141] A relator is an “original source” when he or she either “prior to a public disclosure . . . voluntarily disclosed to the Government the information on which allegations or transactions in a claim are based,” or “has knowledge that is independent of and materially adds to the publicly disclosed allegations or transactions.”[142] Courts have adopted differing interpretations of this statutory language, including as to what standard to apply in determining whether information “materially adds” to publicly disclosed allegations.
In United States ex rel. Forney v. Medtronic, Inc., the Third Circuit was called upon, in the context of a qui tam action, to determine whether Escobar’s interpretation of “materiality” as an element of a fraud claim should govern the analysis of whether information “materially adds” to the allegations for purposes of the public disclosure bar.[143] In United States ex rel. Moore & Co., P.A. v. Majestic Blue Fisheries, LLC,[144] which preceded Escobar, the Third Circuit adopted a broad definition of materiality, holding that the relevant standard in this context is “whether the relator ‘contributes information—distinct from what was publicly disclosed—that adds in a significant way to the essential factual background: the who, what, when, where and how of the events at issue.’”[145] By contrast, the First Circuit in United States ex rel. Winkelman v. CVS Caremark Corp.[146] applied the more “rigorous requirement” for materiality adopted in Escobar, which requires that, “for something to be material, it must be of such importance that it influences the decision maker.”[147]
In Forney, the Third Circuit concluded that the Supreme Court’s interpretation of “materiality” as an element in a fraud claim in Escobar had “little bearing” on the concept of materiality in the entirely different context of the public disclosure bar.[148] Thus, because Escobar did not compel a different interpretation of materiality in this context, the court concluded that it continued to be bound by the Third Circuit’s more lenient standard in Moore.[149] Applying this standard, the court held that the relator qualified as an original source. The relator’s documents included records documenting Medtronic representatives engaging in the challenged activity, including “the names” of individuals involved as well as the “places and times” of the activity.[150] The court rejected the argument that this information was encompassed in the broader and more general allegations in prior public disclosures, holding that the information the relator provided was “detailed, specific, and helpful,” and thus “meaningfully add[ed] to the prior public disclosures.”[151]
F. First-to-File Bar
The FCA’s first-to-file bar “prohibits a person from bringing a ‘related action’ when an FCA suit is pending.”[152] Despite the deceptively simple language, circuits have begun to diverge on how to enforce the bar when the court has already dismissed the earlier, “pending” action.
In United States ex rel. Wood v. Allergan, Inc.,[153] the Second Circuit joined the D.C. Circuit in holding that a relator may not proceed based on an amended complaint if the same relator’s original complaint otherwise would fail under the first-to-file bar. Below, the district court held that, although at least one related complaint against Allergan already existed when the relator filed suit, amending his complaint after the court had dismissed the other, related complaint was sufficient to overcome the bar, because “there were no pending related actions when the complaint was amended.”[154] Focusing on the plain language of the statute, which requires dismissal when a relator “brings” an action while a related action is pending, the Second Circuit disagreed with the district court’s analysis and held that the relator’s action “was incurably flawed from the moment he filed it.”[155] The Second Circuit confirmed, however, that dismissal based on the first-to-file bar should be without prejudice,[156] and that “absent a statute of limitations issue, the relator will be able to re-file her action, without violating the first-to-file bar.”[157]
In the opinion, the Second Circuit also weighed in on another developing circuit split regarding whether a complaint that falls short under Rule 9(b)’s particularity requirement can bar a later-filed complaint under the first-to-file rule. Again agreeing with the D.C. Circuit, the Wood court found no basis to incorporate Rule 9(b)’s particularity requirement into the statute.[158] That a subsequent complaint might be “more detailed” has no bearing on the application of the first-to-file bar under the Second Circuit’s interpretation, and the court thus found that the relator’s allegations were sufficiently related to those in the other, earlier complaint to trigger the bar, even if his allegations were “more detailed” than those previously asserted.[159]
Should the circuit courts continue to diverge on these issues, the resulting split may present opportunities for the Supreme Court to grant certiorari and weigh in on the proper application of the bar. The Court has demonstrated some willingness to address FCA-related issues in recent months when it granted certiorari in United States ex rel. Hunt v. Cochise Consultancy, Inc., 887 F.3d 1081, 1083 (11th Cir. 2018), to address questions regarding the FCA’s statute of limitations provision.
III. PROMOTIONAL ISSUES
This past year saw relatively few developments in the regulation of drug and device promotional activity compared to prior years. Most notably, FDA issued two guidance documents in June 2018 that signaled greater flexibility in FDA’s approach to promotional communications; the guidance greenlighted the transmission of certain information that is not included in FDA-required labels (so long as it is consistent with those labels), as well as economic information about drugs and devices. In a statement about the guidance documents, FDA Commissioner Dr. Scott Gottlieb explained that the guidance is intended to help market participants develop contracts that better reflect the full value of products in the marketplace.[160]
In the meantime, Congress failed to make significant progress toward implementing off-label promotion legislation, and the federal courts did not issue any notable opinions clarifying how drug manufacturers should convey promotional information about their products. FDA enforcement activity increased only slightly relative to last year’s drop in enforcement.
Considering the current administration’s deregulatory position, FDA is likely to continue its scaled back approach to off-label promotion. If the past two years are any indicator, the agency will remain focused on the most flagrant cases of deceptive advertising or when there is a risk to public health, leaving manufacturers some leeway for more expansive promotional activity.
A. FDA Enforcement Activity – Drug Promotion
In 2018, the Office of Prescription Drug Promotion (“OPDP”) issued only two Warning Letters and five Untitled Letters.[161] While this number represents a small uptick from 2017, it still falls far short of the statistics from the years of more aggressive enforcement that preceded the First Amendment litigation.[162] The downward trend may be an indicator that the current administration’s priorities lie elsewhere—at least for the foreseeable future.
Of the seven letters, four involve the omission or minimization of risk information, two concern inaccurate information regarding an unapproved use or product, and one objects to false or misleading claims about efficacy.[163] All of the approved products relate to products with boxed warnings. The letters encompass a range of communications, including conference exhibitions, oral statements by a sales rep, print materials, websites and online vides, and social media.
- In a February 9, 2018 Untitled Letter to Collegium Pharmaceutical, Inc., OPDP asserted that the company’s exhibit booth at an annual pharmacist exhibition made false or misleading representations about an opioid pain reliever because the exhibit failed to communicate accurately the drug’s risks.[164] The letter noted that the exhibit did not include any information regarding the drug’s limitations of use, namely that the drug should be used only as a last resort for treating pain because of its high addiction risks.[165] The letter also emphasized that the omission was particularly concerning considering the severe impact of opioid addiction.[166] Although the exhibit had included a side panel that conveyed some warning information, OPDP noted the small font and found this disclosure to be inadequate.[167]
- Similarly, in a June 19, 2018 Untitled Letter to Pfizer Inc., OPDP objected to a direct-to-consumer promotional video for failing to include any risk information regarding the product, a vaginal ring.[168] OPDP also objected to the testimony of the video’s featured spokesperson, who stated that the drug had worked instantaneously and had not resulted in any side effects in her case.[169] OPDP found that the spokesperson’s claims, even if accurate, misleadingly suggested that other patients would experience similar results.[170]
- In a June 28, 2018 Untitled Letter to Arog Pharmaceuticals, Inc., OPDP asserted that the company violated the FDCA by misbranding an investigational acute myeloid leukemia drug both on the company’s website and at its booth display at an annual hematology meeting.[171] According to FDA, both promotions suggested that the drug was safe and effective, even though FDA had yet to approve the drug for commercial purposes.[172]
- In an August 16, 2018 Untitled Letter to ASCEND Therapeutics US, LLC, OPDP objected to a sell sheet that allegedly made false and misleading claims about the efficacy of ASCEND’s hormonal drug, EstroGel.[173] The sell sheet asserted that EstroGel provides the lowest effective dose of estrogen for treatment of certain moderate to severe menopausal-related symptoms, yet OPDP concluded such claims were not supported by the literature cited because it did not rely on studies comparing the drug to other FDA-approved formulations of estrogen.[174]
- In an October 5, 2018 Warning Letter to MannKind Corporation, OPDP asserted that the company’s Facebook post describing its inhalation powder, Afrezza, as helping “your body work its best and protection from health complications” with “no drama” misleadingly suggested an absence of safety concerns, because it omitted information about potentially life-threatening risks.[175] Although the post contained a small pop-up box with information about the risk of acute bronchospasm in patients with chronic lung disease, OPDP concluded that this did not mitigate the deficiencies of the post.[176]
- In an October 11, 2018 Untitled Letter to Eisai Inc., OPDP objected to oral statements that an Eisai sales representative made to health care professionals at a lunch presentation regarding its Fycompa drug for treatment of partial-onset seizures in epileptic patients 12 years and older.[177] The sales representative purportedly mischaracterized the scope of the new indication for which the company sought FDA-approval in a pending new drug application for pediatric patients and minimized the serious boxed warning risks of behavioral side effects.[178]
- Finally, in an October 22, 2018 Warning Letter to Vanda Pharmaceuticals Inc., OPDP objected to Vanda’s webpage, which it claimed lacked appropriate risk information about its drugs.[179] While the letter acknowledged that the webpage referred visitors to another site that provided the box warnings and safety information, OPDP concluded that this did not mitigate the complete omission of any risk information from the webpage itself.[180]
B. FDA Enforcement Activity – Device Promotion
Enforcement relating to medical device promotion remained quiet as well, with only one Warning Letter to a medical device manufacturer. In addition, CDRH issued seven “It Has Come to Our Attention” Letters, questioning whether marketing claims about certain 510(k)-cleared devices were covered by existing clearances and whether products were being promoted for medical device uses without a required premarket clearance or approval.
- RADLogics, Inc Warning Letter.[181] In its Warning Letter to RADLogics, FDA asserted that the company’s marketing claims exceeded those supported by the 510(k) clearance for the company’s radiological image analysis software application. Citing claims on the company’s website and YouTube video, FDA concluded that the device had been cleared for displaying radiological images for analysis by physicians, but the device was promoted as a “Virtual Resident” that could provide computer-assisted detection and marking of abnormalities in images.
- Letters Regarding Vaginal Treatments. In addition, CDRH issued seven “It Has Come to Our Attention” Letters relating to the promotion of products for vaginal treatments in July, questioning whether marketing claims about the 510(k)-cleared devices were covered by existing clearances and whether products were being promoted for medical device uses without a required premarket clearance or approval. Five of the letters related to laser, radiofrequency, and similar devices that had 510(k) clearances for general uses in dermatologic and surgical applications such as ablation of soft tissue. For example, Cynosure,’s DEKA SmartXide2 Laser System had a 510(k) clearance for incision, excision, ablation, vaporization, and coagulation of body soft tissues in a variety of medical specialties, but FDA objected to the promotion of the device as a “clinically proven laser treatment for the painful symptoms of menopause, including intimacy.” Similarly, FDA objected to “vaginal health restoration” and similar claims for products marketed by Inmode MD Ltd. and Venus Concept, Ltd., noting that these products lacked any device premarket clearance or approval.
In addition to these enforcement letters, in May 2018, FDA filed civil complaints in Florida and California alleging that two stem cell clinics were experimenting on patients with adulterated, misbranded, and unapproved drugs.[182] The first complaint targeted U.S. Stem Cell Clinic LLC of Sunrise, Florida, U.S. Stem Cell, Inc., its Chief Scientific Officer, and its co-owner and Managing Officer. The second was brought against California Stem Cell Treatment Center Inc., Cell Surgical Network Corporation of Rancho Mirage, and two doctors. Despite arguments that FDA approval was not needed because patients receive treatment with their own cells, FDA sought to enjoin these clinics from marketing stem cell therapies that do not have FDA approval. The complaints allege that defendants manufactured unapproved stromal vascular faction (“SVF”) products from patient adipose (fat) tissue. U.S. Stem Cell, Inc. previously faced scrutiny from FDA in August 2017 after several patients reported vision loss after SVF treatments; in a statement, the agency said it was acting because the U.S. Stem Cell Clinic did not address violations outlined in a Warning Letter from August 2017.[183] Finally, the complaints state that recent FDA inspections showed the clinics violated cGMP requirements, including some that could impact the sterility of their products.[184]
Notably, therapies in this space have drawn attention from Commissioner Gottlieb, who has vowed that FDA will crack down on clinics marketing questionable or unsafe treatments. That said, Commissioner Gottlieb also has pledged to ease the path to approval for researchers and companies marketing stem cell therapies and regenerative medicine where treatments are legitimate.[185]
C. FDA’s Promotional Guidance – Drugs & Devices
In keeping with its promise to provide more direction regarding the off-label promotion of drugs and devices, FDA issued two useful guidance documents in June 2018.[186] According to FDA Commissioner Gottlieb, the agency aims to spur a “shift toward innovative, value-based payment arrangements” for medical products.[187] To that end, FDA promulgated guidance to provide clarity to drug manufacturers as they develop communications about their medical products, which in turn will ensure that patients, providers, and insurers have access to a wide range of data that they can harness to negotiate prices.[188]
Communications with Payors. In the first guidance document, FDA authorized companies to share certain information with payors about unapproved products and unapproved uses of approved drugs and devices.[189] The guidance assured drug manufacturers that they will not be subject to FDA enforcement for promoting truthful and non-misleading information that is consistent with FDA-required labeling.[190] According to the guidance, FDA will analyze three factors in determining whether promotional information is consistent with the labeling.
- First, FDA will evaluate whether communications about the product conflict with particular conditions of use in FDA-required labeling.[191]
- Second, FDA will assess whether the promotional information increases the product’s potential for harm to health, relative to the product’s FDA-required labeling information.[192]
- Finally, FDA will consider whether the product’s label enables patients to use the product safely and effectively under the conditions suggested in the product communications.[193]
The guidance provides examples of the kinds of information that FDA would and would not consider consistent.[194] For instance, communications providing context about adverse reactions associated with the product’s use would be consistent with FDA-required labeling.[195] By contrast, information about a product’s use to treat a different disease than indicated in the label would not be consistent.[196] The guidance further states that promotional representations must be “grounded in fact and science and presented with appropriate context.”[197]
Despite these requirements, the guidance still relaxes restrictions on off-label promotion somewhat. While insisting that promotional information have some supporting evidence, the guidance permits drug and device companies to utilize evidence that is insufficient to satisfy FDA approval standards.[198] The guidance also does not explicitly bar inconsistent promotional communications but instead deems them outside the scope of the recommendations.[199] This perhaps suggests that FDA may be marginally more lenient in its assessment of inconsistent promotional information.
Communication of Health Care Economic Information. FDA’s second guidance document also provides drug companies with significant leeway.[200] The guidance concerns the sharing of Health Care Economic Information (“HCEI”) with insurance companies and other payors.[201] HCEI is “any analysis . . . that identifies, measures, or describes the economic consequences . . . of the use of the drug.”[202] In the guidance, FDA authorizes drug manufacturers to disseminate HCEI as long as the information relates to the disease being treated or to a condition in the patient population that the drug’s label indicates can be treated by the drug.[203]
Like the first guidance document, the second explains that FDA will not object to certain communications of information about unapproved products and unapproved uses of approved products.[204] FDA provides several examples, including the information about the anticipated timeline for possible FDA approval, product pricing information, and factual presentations of results from studies.[205] In allowing these communications regarding off-label uses of a product, the guidance emphasizes that payors do not need protection in these exchanges because they are sophisticated audiences that can closely scrutinize a range of HCEI.[206]
Draft Guidance on Presentation of Efficacy Data. Citing research that suggests consumers have improved comprehension and recall of efficacy and risk information when it is presented quantitatively, FDA also issued draft guidance on the presentation of quantitative efficacy and information in direct-to-consumer promotional labeling and advertisements.[207] FDA’s guidance provided recommendations in four categories:
- presenting probability information,
- formatting quantitative efficacy or risk information,
- using visual aids, and
- providing quantitative efficacy or risk information for treatment and control groups.[208]
In the first category, FDA recommended expressing efficacy and risk probabilities in terms of absolute frequencies (such as 57 in 100 or 57%) versus relative frequencies (i.e. 33% reduction in risk) to improve consumer comprehension.[209] As to the second, FDA recommended presenting information in consistent numerical formats and probabilities using whole numbers.[210] Third, FDA endorsed the use of visual aids as a way to help consumers understand efficacy and risk probabilities, so long as they clearly explain the information displayed, are proportionate to the numbers that they are representing, and include representations of both numerators and denominators for numerical ratios.[211] Finally, FDA recommended providing quantitative information for both treatment and control groups to improve consumer understanding of efficacy.[212]
D. Legislative Developments Pertaining to Promotional Issues
There was little to report in the realm of legislative activity relating to drug and device promotional activity in 2018. Federal legislation we have discussed previously in these pages, for example, remained stagnant. The Pharmaceutical Information Exchange Act, which would effectively codify FDA guidance on HCEI by giving drug and device manufacturers greater freedom to share economic information regarding unapproved products, remains a draft bill stalled in the House Energy and Commerce Committee.[213] After holding a consideration and mark-up session, the Subcommittee on Health forwarded the bill to the full House Energy and Commerce committee on January 17, 2018. Since then, the bill has not advanced.[214] Similarly, the draft bill of the Medical Product Communications Act of 2017, which would enable manufacturers to discuss certain off-label information with health care providers, remains in the House Energy and Commerce Subcommittee on Health.[215]
E. Litigation Relating to Promotional Issues
In May 2018, the United States Supreme Court considered two recent FCA off-label promotion cases but ultimately denied certiorari.[216] In 2017, the Fifth and Sixth Circuits had dismissed the cases, United States ex rel. King v. Solvay Pharmaceuticals, Inc. and United States ex rel. Ibanez v. Bristol-Myers Squibb Co., in part, in the former case, because the plaintiffs failed to demonstrate at the summary judgment stage that off-label promotion caused physicians and pharmacies to make false claim submissions to the government.[217] With the Supreme Court refusing to hear either case, for the time being plaintiffs will continue to face challenges in pleading and proving causation in FCA off-label promotion cases.
IV. DEVELOPMENTS IN CGMP REGULATIONS, QUALITY SYSTEM REGULATIONS, AND OTHER MANUFACTURING ISSUES
During 2018, we saw robust enforcement efforts relating to purported cGMP and Quality System regulation (“QSR”) violations. In particular, FDA continues to crack down on manufacturing, quality, and data integrity issues and this year issued a number of notable Warning Letters. These developments, as well as final guidance related to data integrity and compounding pharmacies, among other issues, are discussed below.
A. Notable cGMP Enforcement Activity
In 2018, DOJ announced two notable consent decrees of permanent injunction entered by federal district courts against drug manufacturers to stop the distribution of unapproved, misbranded, and adulterated drugs. This is consistent with DOJ’s publicized enforcement priorities. Notably, in December, Deputy Assistant Attorney General James Burnham gave a speech where he explained that compounding pharmacies “are an increasing focus of enforcement efforts” and “a major enforcement priority” for DOJ and FDA, and highlighted the potential of enforcing cGMP violations as FCA cases.[218] These comments highlight the importance of this recent guidance from FDA for compounding pharmacies.
In April, the U.S. District Court for the Eastern District of Arkansas enjoined Cantrell Drug Company and its co-owner and CEO from distributing adulterated drugs. According to the government’s complaint, the defendants’ drugs were prepared, packed, or held under insanitary conditions and may have been contaminated or rendered injurious to health. By way of background, Cantrell initiated two voluntary recalls of its drug products in 2016 and 2017, both due to a lack of sterility assurance. In addition, during a 2017 inspection, FDA observed that the pharmacy’s own documents memorialized that it had found several types of microorganisms in the air and on surfaces used for sterile processing. The pharmacy then failed to conduct adequate investigations of this microbial contamination. As part of the injunction, the defendants cannot resume manufacturing, processing, or distributing drugs until they submit a remedial plan to FDA providing an independent expert to conduct inspections and ensure the defendants’ compliance with current good manufacturing practice. In reference to this ruling, FDA Commissioner Gottlieb said, “FDA is committed to taking action against compounders who do not comply” with federal requirements.[219]
In June, the U.S. District Court for the Northern District of Mississippi entered an injunction against Delta Pharma, Inc., its President, and its Vice President and Pharmacist in Charge to enjoin further distribution of adulterated drugs. The complaint alleged that during a 2017 inspection, FDA documented the defendants’ failure to establish and follow appropriate written procedures designed to prevent microbiological contamination of sterile drug products. It also noted that the defendants failed to establish an adequate quality control unit with the responsibility to approve or reject all components and investigate any errors that may occur. As part of the permanent injunction, the defendants cannot manufacture, hold, or distribute any drugs until they report to FDA the actions they have taken to correct all deviations from cGMPs.[220]
B. cGMP-Based Warning Letters
FDA’s Office of Manufacturing Quality in the Center for Drug Evaluation and Research (“CDER”) issued 53 Warning Letters in 2018, just off the pace it set in 2017 with 61 Warning Letters. As in 2016 and 2017, FDA continued to focus on issues identified during foreign inspections, sending Warning Letters to companies in China and India, as well as Australia, North and South Korea, and Taiwan. Also consistent with FDA’s activity in 2017, CDER’s 2018 Warning Letters underscore a focus on data integrity (which was also the subject of guidance, discussed below). Data integrity citations and recommendations for “Data Integrity Remediation” cropped up in more than a dozen of the Warning Letters issued by CDER in 2018. CDER most often found these violations in manufacturing facilities in China and India.[221]
We have summarized a few notable Warning Letters below:
- Alchymars ICM SM Private Limited.[222] After a 2017 inspection of the company’s facility in India, FDA issued a February 2018 Warning Letter to Alchymars listing cGMP violations. The alleged violations included data integrity deficiencies; for example, FDA investigators found that Alchymars’s analysts were falsifying laboratory data and neglecting to keep complete records of major equipment maintenance. To address these issues, FDA recommended a comprehensive data integrity remediation. In the Warning Letter, FDA also asserted that Alchymars failed to investigate quality-related complaints, maintain manufacturing buildings and facilities, and provide personnel with adequate clean washing and toilet facilities. Notably, FDA pointed out that it had found similar violations at the company’s facility in 2015, which also resulted in a Warning Letter. FDA admonished Alchymars that the “repeated deficiencies demonstrate that your facility’s oversight and control over the manufacture of drugs is inadequate” and called for a cGMP consultant, a comprehensive investigation into Alchymars’s inaccurate data records, and a current risk assessment.
- Lijiang Yinghua Biochemical and Pharmaceutical Co., Ltd.[223] In April, a Chinese manufacturer received a Warning Letter that cited, among other things, the lack of controls in place to keep staff from altering or deleting electronic data. FDA noted that lab equipment used to generate analytical data had a single username to which all users had access, making it impossible to trace individuals who may have created, modified, or deleted data. FDA also cited the company for failure to maintain complete data from all lab tests, noting that, in response to an FDA request for electronic data, Lijiang answered it had been deleted by accident and was no longer available. Finally, FDA cited the company for failure of its quality unit to wait until testing was complete before approving the release of an active pharmaceutical ingredient (“API”) batch and for failure to adequately investigate deviations, atypical events, complaints, and out-of-specification results. To address these issues, FDA recommended a comprehensive data integrity remediation, including data inaccuracy investigation, a risk assessment, and management strategy. It also noted that many of these violations were repeated after a 2015 FDA inspection and Warning Letter.
- Zhuhai United Laboratories Co., Ltd.[224] In June 2018, FDA cited Zhuhai, a Chinese pharmaceutical company, for multiple violations including quality and data integrity deficiencies. FDA cited Zhuhai’s failure to adequately investigate and document out-of-specification results and to ensure that critical deviations were resolved. For example, in response to below-specification results, Zhuhai simply re-tested and resumed manufacturing after receiving passing results without identifying a laboratory error or otherwise investigating the root cause of the initial results. Its policies stated that using an outlier test (a statistical analysis showing a significant difference between the original value and retest results) could waive the requirement for investigating the cause of atypical results. FDA stated this policy did not comply with good manufacturing practice, as outlier tests may be used only for auxiliary, informational purposes. In addition, FDA noted that Zhuhai did not expand the scope of its review to a larger data set after discovering these significant data lapses. In addition to a data remediation plan, FDA recommended that the company hire a cGMP consultant to correct deviations.
- Boule Medical AB.[225] In October 2018, FDA issued a Warning Letter to a Swedish-based medical device manufacturer of the “Medonic M-Series Hematology Analyzer.” According to the letter, the company failed to meet requirements of the Quality System regulations, which govern quality and consistency in manufacturing of medical devices. FDA cited the company for six separate violations, principally related to failures to maintain effective policies, procedures, and processes.
- StemGenex Biologic Laboratories, LLC.[226] Also in October 2018, FDA issued a Warning Letter to StemGenex, a San Diego–based stem cell company, for a litany of alleged cGMP violations (as well as promotional issues). Given that many of FDA’s cGMP actions originate overseas, the letter targeting a U.S.-based facility is comparatively rare, although FDA scrutiny of companies exploring stem cell technology is not uncommon in recent history. FDA cited StemGenex for “unvalidated manufacturing processes, uncontrolled environment, lack of control of components used in production, [] and lack of sufficient and validated product testing.” According to FDA, these shortcomings posed “a significant risk that [the company’s] product may be contaminated with microorganisms or have other serious product quality defects.”
C. Quality System Regulation
FDA pursued QSR violations aggressively in 2018, including against foreign companies. Of the 25 device-related Warning Letters, 19 were issued for QSR violations. A few representative Warning Letters are summarized below:
- Dexcowin Co., Ltd.[227] After a 2017 inspection of the company’s facility in South Korea, FDA issues a February 2018 Warning Letter to Dexcowin, a portable dental diagnostic X-ray device manufacturer, listing QSR violations. The alleged violations included a failure to establish and maintain adequate procedures for implementing corrective and preventive action (“CAPA”), as required by 21 CFR 820.100(a), failure to maintain required data, and training procedure deficiencies. FDA noted that “[g]iven the serious nature of the violations,” FDA was taking steps to refuse entry of these devices into the United States “until these violations are corrected.”
- US Vascular, LLC.[228] In June, FDA issued a Warning Letter to the manufacturer of Class II devices used in vascular pathology for alleged QSR violations—including failure to establish required procedures for design control and change; receiving, reviewing, and evaluating complaints; CAPA; and document and product control. FDA also alleged MDR violations in its Warning Letter FDA admonished the company for its repeat violations, noting that 11 of 12 citations “were repeat citations from the April 2017 inspection,” and 8 citations “were also repeated from the March 2016 inspection.” FDA informed the company that because it had “not provided any objective evidence of corrections to date,” the adequacy of the company’s response with respect to several of the violations could not be determined.
- Anigan, Inc.[229] In a July Warning Letter to a manufacturer of reusable menstrual cups, FDA alleged several QSR violations, such as inadequate procedures for design control and validation, finished device acceptance, and quality audits. FDA admonished the company for failing to conduct risk analysis for either model of the company’s menstrual cups. FDA also alleged that management with executive responsibility at the company had “not reviewed the suitability and effectiveness of the quality system.” FDA recommended that the company review the relevant regulatory requirements and promptly correct its violations.
- Becton Dickinson Medical Systems.[230] In September, FDA issued a Warning Letter to a manufacturer of lock flush syringes for alleged QSR violations. FDA cited the company for five separate violations, principally related to failures to maintain effective procedures and processes. Among the issues, FDA alleged that the company failed to adequately establish procedures “to control environmental conditions” and “to prevent contamination of equipment or product.” Although FDA determined that the company’s response “appear[ed] to be adequate,” a follow-up inspection would be required to assess the ongoing corrective actions.
D. cGMP Rulemaking and Guidance Activity
In 2018, FDA also issued several notable guidance documents related to cGMP issues, including the following:
- Repackaging Biologics. In January, FDA issued final guidance related to cGMP compliance; the document is entitled Mixing, Diluting, or Repackaging Biological Products Outside the Scope of an Approved Biologics License Application.[231] It replaces (and largely replicates) 2017 draft guidance of the same name. The guidance document details the conditions under which state-licensed pharmacies, federal facilities, or outsourcing facilities can mix, dilute, and repackage biologics. According to FDA, “diluting or mixing a biological product with other components, or repackaging a biological product by removing it from its approved container-closure system and transferring it to another container-closure system, is, in the absence of manufacturing controls, highly likely to affect the safety and/or effectiveness of the biological product.”[232] FDA recognized, however, that in certain circumstances, it is appropriate to mix, dilute, or repackage a biological product to meet the needs of a specific patient (e.g., in the pediatric care context).[233] Although any biological product that is mixed, diluted, or repackaged such that it falls outside the scope of an approved biologics license application is an “unlicensed biological product,” this guidance details the appropriate conditions under which these steps can be taken without risk of FDA action. Notably, this document contains additional requirements for outsourcing facilities to pass certain tests and to notify customers of any failing results in order to avoid FDA action.[234]
- Compounding Pharmacies. In December, FDA issued revised, draft guidance for compounding pharmacies: Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act.[235] The draft guidance, which revises 2014 guidance, is specific to outsourcing facilities and is intended to fill a gap until FDA “promulgate[s] more specific cGMP regulations for outsourcing facilities.” In that context, the draft guidance focuses on cGMP requirements intended to ensure sterility of the drug manufacturing process, and safety issues that pose the “highest risk,” including ensuring the appropriate potency and labeling for compounded drugs. The draft guidance makes specific recommendations for quality assurance, facility design, manufacturing controls, and other topics.
- Data Integrity. FDA also issued final guidance related to Data Integrity and Compliance With Drug CGMP.[236] The guidance emphasizes the importance of “reliable and accurate” data, but allows for “flexible and risk-based strategies to prevent and detect data integrity issues.” Notably, the guidance suggests that “management with executive responsibility” must “create a quality culture where employees understand that data integrity is an organizational core value and employees are encouraged to identify and promptly report data integrity issues,” and warns that data integrity issues in manufacturing processes can lead to Warning Letters, import alerts, and consent decrees. FDA Commissioner Gottlieb described the guidance as “one part of [FDA’s] multi-layered approach to ensuring the integrity of data,” which also includes the “pre-approval inspection process” and “thorough assessments of applications prior to approval and when companies submit information about changes to their manufacturing processes.”[237]
V. ANTI-KICKBACK STATUTE
As outlined above in the summary of recent DOJ enforcement activity, DOJ routinely invokes the AKS in bringing FCA claims against health care companies and drug and device makers in particular. Below, we outline several notable developments in AKS-related case law and guidance. We also address a developing story regarding the administration’s interest in rolling back safe harbor protection for drug rebates, one prong of its “Blueprint” to decrease drug prices in the United States.
A. AKS-Related Case Law
Although relatively quiet, 2018 delivered several notable cases involving pharmaceutical companies and medical device makers.
Causation in AKS-Premised FCA Cases. In January, the Third Circuit affirmed the grant of summary judgment in favor of a pharmaceutical company accused of FCA and AKS violations in connection with its charitable donations.[238] In United States ex rel. Greenfield v. Medco Health Solutions, Inc., the relator alleged that a specialty pharmacy, Accredo Health Group, which delivered clotting medication to patients at home and provided nursing assistance for patients with hemophilia, made donations to two charities that then recommended the company to hemophilia patients. The U.S. District Court for the District of New Jersey denied the relator’s motion for summary judgment and granted the company’s, finding that the relator was unable to show that the charities’ referral of several federally insured patients resulted from the pharmaceutical company’s charitable contributions.
On appeal, the relator argued that the district court erred in requiring a direct link between the contributions and the referrals, and the government filed an amicus brief in support of neither party contending that the Court erred in requiring the relator to prove that patients chose the company because of the charities’ recommendations. The government also asserted that, for purposes of establishing an FCA violation, the relator need only demonstrate that a claim that sought reimbursement for medical care was provided in violation of the AKS. For its part, the specialty pharmacy maintained that the district court correctly framed the relator’s burden of establishing an FCA breach in requiring the relator to point to some evidence that the pharmacy received referrals for federally insured patients “because of” its allegedly improper donations.
Applying the post-Patient Protection and Affordable Care Act (“PPACA”) version of the AKS, which provides that “a claim that includes items or services resulting from a violation of [the AKS] constitutes a false or fraudulent claim for purposes of [the FCA],”[239] the Third Circuit ultimately affirmed the district court’s grant of summary judgment in favor of the specialty pharmacy. The Third Circuit held that a relator, at minimum, must show that at least one of the patients for whom the company submitted reimbursement claims was exposed to a referral from a charity that received a donation. Of note, the Third Circuit disagreed with both parties as to the necessary standard, concluding that there must be “some connection between a kickback and a subsequent reimbursement claim.” The Third Circuit explained further that “[i]t is not enough . . . to show temporal proximity between [the company’s] alleged kickback plot and the submission of claims for reimbursement[,]” but it also is “too exacting to . . . require[] a relator to prove that federal beneficiaries would not have used the relevant services absent the alleged kickback scheme.”[240]
Escobar and AKS-Based FCA Cases. In United States ex rel. Medrano et al. v. Diabetic Care RX, LLC, a magistrate judge for the U.S. District Court for the Southern District of Florida recommended dismissal of the government’s FCA claims against compounding pharmacy, Diabetic Care RX d/b/a Patient Care America (“PCA”), and its private equity fund owner, Riordan, Lewis & Haden, Inc., for alleged AKS violations.[241] After intervening in the case, the government alleged that PCA paid kickbacks to marketers through profit-sharing arrangements in exchange for prescription referrals, and that the marketers covered patients’ copayments to induce those patients to accept the defendant’s compounded medications.[242] The government also alleged that PCA hid its contribution to some of the prescription copayments through a “sham charitable organization.”[243]
The magistrate judge held that the argument that PCA’s reimbursement claims were “tainted” by the alleged kickbacks failed to state a valid FCA claim under either the express or implied certification theory. In so holding, the magistrate judge explained that the representative claims cited in the complaint failed to identify any “specific representations” made to Tricare when submitting those claims,[244] and that the complaint only cited certifications related to a provider agreement between PCA and the Tricare pharmacy benefits manager that pre-dated submission of the allegedly false claims.[245] The district court has yet to weigh in on the magistrate judge’s recommendations.
The AKS’s Exemption for Payments to Employees. In deciding Carrel v. AIDS Healthcare Foundation, Inc. this past August, the Eleventh Circuit examined the AKS provision exempting payments to employees from the statute’s reach.[246] As noted above, the relators claimed that the foundation improperly paid bonuses to its employees for referring patients with HIV/AIDS to other services provided by the foundation. But the Eleventh Circuit concluded that the AKS employee exemption provision—which immunizes payments from employers to their employees “for employment in the provision of covered items or services”[247]—applied to the referral payments.[248] The court reasoned that the plain language of the Ryan White Act, 42 U.S.C. § 300ff-51(e)(2), classifies the referral of patients with HIV/AIDS to entities funded under the Act as “a standalone compensable ‘service,’” and thus the payments were tied to employment in the provision of a covered service.[249] Emphasizing that the “relevant statutes say nothing to forbid payment on a per-capita basis or to require nondiscriminatory referrals to any available healthcare provider,”[250] the court found that the referrals to the foundation thus qualified as a “covered . . . service[]” under the statutory exemption, and that the foundation’s payments to its own employees for those referrals did not violate the AKS.[251]
The One Purpose Test. In United States ex rel. Derrick v. Roche Diagnostics Corp., the U.S. District Court for the Northern District of Illinois allowed a relator’s AKS-based FCA claims to move forward after deciding that the complaint sufficiently alleged that the payment claims were “tainted” by underlying AKS violations.[252] According to the complaint, Roche contracted with the insurance company, Humana, to list its glucose monitoring products on Humana’s Medicare formularies but later discovered that it had overpaid certain rebates to Humana.[253] The relator alleged that, after Humana notified Roche that it would be removing Roche’s products from the formularies, Roche forgave the majority of that debt in exchange for remaining on Humana’s formularies and excluding competing brand products from those formularies.[254] In denying the defendants’ motions to dismiss, the district court explained that the relator need not identify a specific claim for payment that “included items or services resulting from an AKS violation,” as Humana’s Medicare contracts required monthly payment claims covering the products at issue, and the allegations “necessarily le[d] to the conclusion that Humana presented claims to CMS that were tainted by the alleged fraud.”[255] The court further explained that, even if the debt forgiveness arrangement “had a legitimate business purpose,” it could constitute “illegal remuneration for AKS purposes if one reason for it was to compensate[] past or induce[] future referrals.”[256]
Addressing the Remuneration Element. In State v. MedImmune, Inc.,[257] the U.S. District Court for the Southern District of New York held that the State of New York adequately pled that “the sharing of [personal health information (“PHI”)] with specialty pharmacies could plausibly constitute ‘remuneration’ in violation of the federal anti-kickback statute” so as to support the State’s claim that MedImmune caused a specialty pharmacy to submit false billing certifications in violation of the New York State False Claims Act.[258] Although the State alleged that MedImmune sales personnel would “curry favor” with hospital administrators in exchange for access to confidential PHI, the crux of the State’s claims centered on whether MedImmune violated the AKS by passing that PHI on to the specialty pharmacy as “leads” to use in pursuing prime candidates for MedImmune’s neonatal respiratory drug.[259] The ruling joins the growing number of district courts taking a broad approach to the definition of “remuneration” under the AKS at the motion-to-dismiss stage.
B. Legal Challenges to AKS-Based Theories
In December, DOJ moved to dismiss 11 qui tam FCA actions brought by Health Choice Group, LLC against multiple pharmaceutical companies.[260] Rejecting allegations that the defendants violated the AKS by providing prior authorization assistance and arranging for nurses to educate patients on proper administration of newly prescribed medicines, the government argued that the relators’ allegations “lack[ed] sufficient factual and legal support” and would be contrary to previous guidance issued by HHS OIG on the provision of patient educational materials and informational programs.[261]
The government emphasized that the right for a qui tam plaintiff to proceed in the absence of government intervention “is not absolute,” and it moved for dismissal given the need to “preserv[e] scarce government resources and protect[] important policy prerogatives of the federal government’s healthcare programs.”[262] Specifically, the government maintained that the “relators should not be permitted to indiscriminately advance claims on behalf of the government against an entire industry that would undermine common industry practices the federal government has determined are . . . appropriate and beneficial to federal healthcare programs and their beneficiaries,” including the “provision of educational information and instruction to patients” once a physician “has appropriately prescribed a medication.”[263] Although the fact that the relator was an entity created to bring FCA qui tam suits may have impacted DOJ’s approach to these cases, the government’s effort to dismiss these actions—like the Campie case discussed above—may signal a potential shift in enforcement policy under the parameters outlined in the updated Justice Manual and Granston Memo.
Whereas DOJ opted to challenge Health Choice Group’s AKS theories, both DOJ and HHS OIG have challenged patient assistance programs under kickback-based theories over the past few years. Indeed, in our 2017 Year-End Update, we discussed HHS OIG’s increased scrutiny of patient assistance programs, including the unprecedented rescission of guidance previously issued to an industry-funded charity that operated a patient assistance program due to alleged breaches of two commitments related to donor independence.[264] We also discussed the $210 million settlement in December with another patient assistance charity for similar violations.[265]
In response to this enforcement and regulatory environment, a patient assistance charity filed a lawsuit in January challenging HHS OIG advisory opinions regarding interactions with pharmaceutical industry donors on First Amendment grounds.[266] The charity’s complaint alleges that recent revisions to an advisory opinion infringe on the charity’s freedom of speech. According to the charity, it had relied on the advisory opinion since 2002, and the revisions came after a three-year negotiation period with HHS OIG following its release of a special bulletin in 2014 that warned about the risk of fraud and abuse at patient assistance charities. Among the revisions are required commitments not to solicit donor suggestions as to the selection of which diseases to address through funding decisions, not to let donors influence the distribution of funding, and not to fund disease therapies at the request of donors that sell treatments for those diseases. At its core, the complaint alleges that these required commitments amount to “new and oppressive restrictions that cripple [the charity’s] ability to carry out its charitable efforts.”[267]
The court recently granted the government’s motion for limited discovery into whether the charity waived its First Amendment rights by agreeing to the advisory opinion’s terms and “whether the interests promoted by enforcement of the speech-related provisions of OIG’s advisory opinion outweigh any public policy harms that would result from enforcement.”[268] As the lawsuit progresses, it could develop into a case with serious implications for the government’s ability to control truthful speech regarding federally reimbursable drugs.
C. Rulemaking
HHS made headlines in 2018 with a proposed rule that would roll back a significant AKS safe harbor. In mid-July, HHS submitted a proposed rule for review and clearance to the Office of Management and Budget (“OMB”) titled “Removal Of Safe Harbor Protection for Rebates to Plans or PBMs Involving Prescription Pharmaceuticals and Creation of New Safe Harbor Protection.”[269] Although the substance of the rule was not publicly available, the title made clear that HHS was considering potentially allowing AKS enforcement actions against either drug manufacturers, Pharmacy Benefit Managers (“PBMs”), or both, when manufacturers pay PBMs rebates for services such as exclusively covering a manufacturer’s products or favoring such products through a prescription benefit plan. On January 31, 2019, HHS ultimately issued a proposed rule that would amend the definition of “discount” to eliminate the safe harbor covering rebates paid to Part D insurers, Medicaid managed care organizations, and PBMs.[270] The rule change would not impact discounts to distributors, providers, and pharmacies. With a proposed effective date of January 1, 2020, the rule also would add two new safe harbors protecting discounts provided directly to patients “at the point of sale,” if those discounts meet certain criteria, as well as “low risk” fixed fee arrangements for services provided by PBMs to manufacturers, if pursuant to written agreements that reflect fair market value and are not tied to the volume or value of generated business.
Such a shift in safe harbor policy would be consistent with the administration’s “American Patients First: The Trump Administration Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs” initiative discussed below. HHS Secretary Alex Azar also has hailed the proposal as having “the potential to be the most significant change in how Americans’ drugs are priced at the pharmacy counter, ever,” and has praised its potential impact on transparency in the drug markets.[271]
In August, HHS also sought comments on the proposal to expand certain safe harbors and exceptions to the Civil Monetary Penalty’s (“CMP”) definition of “remuneration” in an effort to “foster arrangements that would promote care coordination and advance the delivery of value-based care.”[272] Recognizing the “broad reach of the [AKS] and beneficiary inducements CMP as a potential impediment to beneficial arrangements that would advance coordinated care,”[273] HHS solicited feedback on potential “care coordination,” “value-based arrangements,” “alternative payment models,” “innovative technology” arrangements, and “other novel financial arrangements,” as well as “the types of incentives providers, suppliers, and others are interested in providing to beneficiaries” to improve quality of care, care coordination, and patient engagement.[274] HHS also sought input on the benefits and risks of “relieving or eliminating beneficiary cost-sharing obligations,” proposals for donated or subsidized “cybersecurity-related items and services” for providers, and implementation of other recently passed legislation.[275] HHS received over 350 comments before the end of the comment period in October. The AKS consistently has faced industry critique that its provisions are outdated and unable to keep pace with innovative technologies and shifting reimbursement trends that have moved from fee-for-service systems to more outcome- and value-based models. The allowance for new value-based pricing, collaboration, and risk-sharing arrangements may provide opportunities to align incentives among providers and market participants, as well as opportunities to leverage the specialized knowledge and experience of drug and device manufacturers to identify potential avenues for coordinating and optimizing care in a more cost-effective and patient-friendly manner.
Whether HHS incorporates the industry’s feedback remains to be seen, but we will keep you apprised of any developments related to these proposed policy changes in future updates.
D. Legislative Developments
Under the Physician Payments Sunshine Act provisions of the PPACA, drug and device manufacturers must report annually payments made to physicians and teaching hospitals.[276] To increase transparency and accountability, the “Open Payments” database catalogues information about those payments in a searchable online repository available to the public. One legislative development in 2018 will impact such future reporting requirements.
The SUPPORT for Patients and Communities Act noted above expanded these reporting requirements for pharmaceutical and medical device manufacturers. As of January 1, 2022, manufacturers also must report through the Open Payments system any transfer of value to physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, and certified nurse midwives.[277]
E. HHS OIG Guidance
HHS OIG issued a number of advisory opinions discussing the AKS in 2018, although only three pertained to drug and device manufacturers.
In May, HHS OIG endorsed an arrangement under which a company that manufactures and distributes medical devices and pharmaceutical products provides free samples of ostomy products to patients and then contracts with a third party to conduct customer satisfaction surveys regarding the sample products.[278] HHS OIG gave significant weight to the fact that the company informs patients that they are not permitted to bill third-party payors for sample products, meaning federal health care programs should not incur any costs associated with the arrangement.[279] Further, HHS OIG noted that the contractor conducting the surveys does not sell the medical device company’s products and is not compensated in connection with future sales of such products.[280]
On September 10, HHS OIG evaluated a surgical device and wound care product manufacturer’s proposed warranty program for a suite of three products used in joint replacements.[281] Under the program, the manufacturer would refund hospitals for the aggregate purchase price of the product suite without regard to insurance status or payor if (1) an inpatient received all three products within the “suite,” (2) the patient was readmitted to the same hospital as an inpatient within 90 days due to a surgical site infection or for a revision to the implanted system, and (3) each of the manufacturer’s products was used in accordance with its instructions for use and other labeling, and the hospital certifies that the readmission was due to a failure of the product “to perform as expected.”[282] Although HHS OIG concluded that the safe harbor for warranties, 42 C.F.R. § 1001.952(g), does not apply to bundled items, it concluded that the proposed arrangement “pose[d] a sufficiently low risk of fraud and abuse under the [AKS].”[283]
Specifically, because none of the products in the suite is separately reimbursable, HHS OIG found that the risk of “overutilization,” “inappropriate” product use, and increased Medicare costs was low given the incentive for hospitals to choose the best product.[284] HHS OIG also emphasized the increased transparency and oversight stemming from required cost reporting and the manufacturer’s commitment to meet all seller obligations under the warranties safe harbor, which would “put hospitals on notice of their obligation to appropriately report any refund they obtained.”[285] Beyond recognizing the “innovative and potentially beneficial” qualities of the warranty program in reducing readmission through encouraged use of the product suite, HHS OIG also cited proposed hospital certifications certifying that physicians would remain responsible for medical decisions, as well as the lack of exclusivity requirements or other quotas, minimums, and volume- or referral-based eligibility criteria.[286]
On November 13, HHS OIG concluded that a drug company’s proposal to provide free doses of its drug to hospitals to treat inpatients with a particular condition could constitute improper remuneration under the AKS.[287] The company proposed to stock the drug at participating hospitals on a consignment basis, under which physicians seeking to prescribe the drug would submit a referral to the drug’s reimbursement hub and initiate therapy using the free vial.[288] The reimbursement hub would provide additional free vials if needed for inpatient use and would provide the same for outpatients unable to secure insurance coverage for the drug.[289]
Considering publicly available information outside of the materials submitted by the drug company, HHS OIG provided several reasons for its decision, including that:
- the proposed arrangement “would relieve a hospital of a significant financial obligation that [it] otherwise would incur” in light of the “substantial price increases” for the drug in recent years;
- “no portion of the significant savings [to the hospital] . . . would be passed on to the Federal health care programs” since the drug is not separately reimbursable in the inpatient setting;
- the proposal “could function as a seeding arrangement,” because insurers (including federal health care programs) may end up paying for the drug when insured patients need to finish the treatment on an outpatient basis, and because providing the free drug to inpatients “facilitates [the] high price for the [d]rug’s other indications”;
- it “could result in steering or unfair competition” by “influenc[ing] prescribers to consider the [d]rug as a first option”;
- providing the drug on the proposed consignment basis actually would eliminate two of the barriers cited by the drug company (i.e., immediate access and unwillingness to bear excess inventory risks); and
- certifications that the free vials would not be contingent on future purchases “ring[] hollow,” as “receipt of the free vial would be contingent on future purchases of the [d]rug” for any outpatients who have insurance coverage and must purchase the drug to avoid potential adverse medical consequences from halting the treatment they began for free on an inpatient basis.[290]
VI. DRUG PRICING
Signaling a reinvigorated focus on drug pricing, the Trump Administration announced the “American Patients First: The Trump Administration Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs” initiative (the “Blueprint”) in May 2018, which aims to lower prices for consumers through a combination of increasing competition among drug manufacturers, strengthening Medicare’s negotiating power, and lowering Medicare and Medicaid list prices.[291]
The announcement of the Blueprint spurred several related HHS and FDA initiatives throughout 2018, and the administration’s focus on drug pricing shows no signs of abating as we transition into 2019. Further, the House Committee on Oversight and Reform already has “launched one of the most wide-ranging investigations in decades into the prescription drug industry’s pricing practices” with the goal of “determin[ing] why drug companies are increasing prices so dramatically, how drug companies are using the proceeds, and what steps can be taken to reduce prescription drug prices.”[292] The investigation will begin with requests for detailed information and documents regarding pricing practices from 12 drug companies, and the Committee has pledged to “hold its first of several hearings in the coming weeks.”[293]
Below, we address several other notable moves by HHS and FDA on the drug pricing front.
A. Rulemaking
In support of the Blueprint’s efforts to increase pricing competition and negotiations and lower list prices, CMS proposed several rules in the latter half of the year that could impact drug pricing in the future.
In October, CMS proposed a rule that would require direct-to-consumer (“DTC”) television ads for certain Medicare- and Medicaid-reimbursable prescription drugs and biological products to display the list price of the drug or product.[294] Although CMS touted the ability of consumers to “price shop” based on the pricing information that would be included in the ads,[295] it is unclear how the market would react to such pricing information, as most customers do not pay list prices for prescription drugs or biological products. The proposed rule also clarifies that the Secretary of HHS would maintain an annual “public list” of all drugs and biological products that it identifies as “in violation of th[e] rule” but indicates that the “primary enforcement mechanism” would be “the threat of private actions” under the Lanham Act’s prohibitions on false or misleading advertising.[296] The comment period ended on December 17, 2018. If the proposed rule moves forward, it may trigger challenges to CMS’s rulemaking authority and First Amendment objections.
Also in October, CMS issued an advanced notice of proposed rulemaking to solicit public comments on a new “International Pricing Index” payment model, which aims to “phas[e] down” the prices that Medicare Part B pays for “selected” physician-administered drugs to align more closely with international prices in “economically-similar countries.”[297] The notice highlights several shortcomings with the current system, including the use of a six percent add-on payment keyed to the price of the drug rather than a “set payment reflecting the service being performed,” as well as the financial risk placed on providers under the traditional “buy-and-bill system.”[298] In addition to phasing down the Medicare payment amount for selected Part B drugs, the proposed model would “[a]llow [approved] private-sector vendors to negotiate prices for drugs, take title to drugs, and compete for physician and hospital business” and would “[p]ay physicians and hospitals [outpatient departments] the add-on based on a set payment amount structure.”[299] The comments period for the proposed rule closed on December 31, 2018.
In November, CMS also proposed a rule that would introduce changes to Medicare Part D and Medicare Advantage drug plans in an effort to support drug price negotiations and reduce out-of-pocket costs.[300] The rule proposes to revise certain protections that currently require Medicare Part D plans to cover every FDA-approved drug in six specially designated therapy classes. The new rule would create certain exceptions to the protected class policy, under which Part D sponsors could use prior authorization and step therapy for protected class drugs, exclude any protected class drugs that are updated formulations of existing protected drugs (even if the old formulation is no longer available), and exclude a protected class drug from its formulary when the price for that drug rises faster than inflation over a designated period.[301] Consistent with FDA guidance described below on non-protected class drugs, the proposed rule also would allow Part D sponsors to introduce “indication-based formulary design and utilization management” for protected class drugs, allowing them to create drug formularies based on self-chosen disease indications.[302] Under the proposal, Part D sponsors also must implement a Real-Time Benefit Tool by January 1, 2020, alerting prescribers to lower-cost therapy alternatives under the patient’s plan benefits,[303] and the plans’ Explanation of Benefits must include information about negotiated price changes and lower-cost therapeutic alternatives for beneficiaries.[304]
B. Related Guidance
In August, CMS issued two guidance documents with potential implications for drug pricing. Rescinding previous guidance, CMS issued a memorandum on August 7, 2018, permitting Medicare Advantage (“MA”) plans to use step therapy for Part B drugs as of January 1, 2019, “as part of a patient-centered care coordination program.”[305] Step therapy is a form of prior authorization that begins patients on medication “with the most preferred drug therapy” for a particular medical condition and “progresses to other therapies only if necessary,” allowing some patients to begin treatment with a more cost-effective medication before moving to more expensive options if the first proves ineffective.[306] In issuing the guidance, CMS emphasized that allowing step therapy for Part B drugs “will help achieve the goal of lower drug prices while maintaining access to covered services and drugs for beneficiaries,”[307] and it clarified that an MA plan opting for step therapy Part B drugs must “offer beneficiaries an opportunity to participate in drug management care coordination activities”[308] and should pass on savings and encourage enrollees to participate in such programs by “offering rewards in exchange for enrollee participation” in accordance with applicable laws.[309] MA plans that choose to offer Part B step therapy programs also must adhere to certain disclosure and reporting obligations outlined in the guidance.
In another reversal, CMS issued a memorandum on August 29, 2018, allowing Medicare Part D plans to tailor on-formulary coverage by drug indication.[310] Such “indication-based formulary design” is allowed in the private sector, but Part D plans currently must cover every FDA-approved indication for drugs included on their formularies, which can limit their ability to negotiate discounts.[311] CMS hopes that the flexibility of including different drugs for different indications will “provide additional negotiating leverage” on drug prices and lead to greater diversity of drug choices and coverage plans.[312] The memorandum also clarifies that Part D sponsors must include a “therapeutically similar drug on formulary” for any indications for which it plans to limit coverage of a particular drug.[313] In addition, Part D plan sponsors must update beneficiary materials to communicate any indication limitations.[314]
FDA also took action aimed at increasing the availability of nonprescription drugs and lowering drug prices, including through issuing draft guidance that outlines potential approaches to consider when demonstrating safety and effectiveness for nonprescription drug products,[315] issuing 63 product-specific guidances to promote generic drug access and drug price competition,[316] establishing a working group to examine the possibility of “import[ing] prescription drugs from other countries in the event of a dramatic price increase” for drugs with only one manufacturer and without patents or exclusivities,[317] and releasing a Biosimilar Action Plan to improve the development and approval process and “encourag[e] innovation and competition among biologics and the development of biosimilars.”[318]
VII. MEDICAL DEVICES
In 2018, FDA published multiple guidance documents and rulemakings aimed at making device clearance approval processes more efficient, fostering innovation, and promoting cybersecurity and device safety. In this section, we provide an overview of CDRH’s strategic priorities, the significant reforms of the device premarket review programs, the Medical Device Safety Action Plan, other guidance developments, and device-related enforcement activity.
A. CDRH Strategic Priorities for 2018 to 2020
CDRH’s strategic priorities for 2018 to 2020 aim to ensure that patients in the United States are the first in the world to have access to cutting-edge, safe, and effective devices.[319] CDRH’s ultimate goal is for more than 50% of manufacturers of novel technologies to “intend to bring their devices to the U.S. first or in parallel with other major markets by December 31, 2020.”[320] In order to achieve this broad goal, CDRH will focus on three strategic priorities, namely (1) improving employee engagement, (2) increasing simplicity, and (3) building collaborative communities.[321] The report suggests FDA’s awareness that the regulatory process must be made less burdensome in order to ensure that the United States remains a country that fosters innovation.
Finding that “[its] issues are often complex,” and “[its] solutions and processes do not necessarily have to be,”[322] CDRH is making it a priority to promote simplicity by streamlining its policies, processes, and programs to more quickly and efficiently achieve CDRH’s goal and vision. As stated in the report, this may involve “removing unnecessary burdens [CDRH] impose[s] on [itself], such as through cumbersome processes, vague policies, and out of date information technology systems.”[323]
With respect to collaborative communities, CDRH states that the American public is best served when stakeholders, including CDRH, work together to proactively solve problems. To that end, CDRH aims to establish at least ten new Collaborative Communities by December 31, 2020, which will allow consumers to take a more active role in “the advancement of smart regulation.”[324] One example proposed by CDRH included collaborative forums for next generation sequencing tests, in which clinicians, test developers, patients, and other professionals would work together to assess the evolving science and recommend when there is adequate evidence supporting the association between particular genetic variants and specific diseases or conditions.[325]
B. Reforms of FDA’s Premarket Review Program
In 2018, FDA focused on reinvigorating and streamlining the premarket review of medical devices, particularly the 510(k) program, which remains the most common premarket pathway for devices. Eighty-two percent of the devices cleared or approved in 2017 were through a 510(k). These activities followed FDA Commissioner Gottlieb’s ongoing goals to modernize 510(k) review, permitting increased flexibility and facilitating a streamlined process that could potentially accelerate the rate at which new innovations are brought to market.[326]
Older Predicate Devices. On November 26, 2018, FDA announced that it would seek to reduce the reliance on older predicate devices (e.g., predicates that are less than ten years old) and “to drive innovators toward reliance on more modern predicate devices or objective performance criteria.”[327] FDA also is considering making public those 510(k)-cleared devices that demonstrated substantial equivalence to older predicates. While emphasizing that the agency did not believe that devices that rely on older predicates are unsafe, FDA’s view is “that encouraging product developers to use more modern predicates would give patients and their doctors a choice among older and newer versions of a type of device, promote greater competition to adopt modern features that improve safety and performance, and help make sure that newer devices reflect more modern technology and standards that can improve patient care and outcomes.”[328]
New Safety and Performance Based Pathway. Last month, FDA finalized guidance on the new “Safety and Performance Based Pathway,” which expands on the concepts underlying the Abbreviated 510(k) pathway. The new pathway relies on modern performance-based criteria and current technological principles to demonstrate substantial equivalence, replacing the direct, head-to-head comparisons to older predicates that have been a feature of traditional 510(k)s.[329] Devices eligible for this pathway must satisfy all of the following factors: they have indications for use and technological characteristics that do not raise different questions of safety and effectiveness than the identified predicate, and the predicate is within the scope of the list of eligible device types; the performance criteria align with the performance of one or more legally marketed devices of the same type as the new device; and the new device meets all the FDA-identified performance criteria. All performance criteria will be published in FDA guidance, and FDA intends to publish guidance on the performance criteria for each device type and recommended testing methods.
Expanded Special 510(k) Program. FDA has initiated a pilot program to expand the scope of the Special 510(k) Program, as described in draft guidance issued on September 28, 2018.[330] The Special 510(k) Program is a streamlined review process for minor, well-defined changes made to already cleared devices. Originally, the program did not include changes affecting a device’s intended use or fundamental scientific technology, but the draft guidance makes these changes potentially eligible for this faster review.
Improvements for the De Novo Classification Process. FDA anticipates that the reforms to the 510(k) pathway may result in an uptick in De Novo classifications. In December 2018, FDA proposed a new rule establishing detailed procedures and criteria for the De Novo classification process to expand the use of the De Novo pathway by providing structure and clarity to applicants seeking this classification for novel, low to moderate risk devices under the FDCA, 21 U.S.C. 360c(f)(2).[331] If finalized, the rule would become part of the Medical Device Classification Procedures under 21 C.F.R. Part 860.[332]
The proposed rule defines the term “De Novo request” and includes guidance on requirements related to the format and content of De Novo requests, as well as procedures for review and criteria for accepting, granting, declining, and withdrawing them. It reiterates that an applicant may submit a De Novo request after submitting a 510(k) and receiving a not substantially equivalent (“NSE”) determination, or before submitting the 510(k) if the applicant “determines that there is no legally marketed device upon which to base a determination of substantial equivalence.”[333] The proposed rule specifies that requests must include “administrative information, regulatory history, device description, classification summary information, benefits and risks of device use, and performance data to demonstrate reasonable assurance of safety and effectiveness.”[334] FDA may refuse to accept or decline a De Novo request if the request is incomplete or the device is ineligible for such a classification.[335]
Other Notable Guidance Relating to Device Premarket Review.
- The Q-Submission Program – draft guidance. The Q-Submission Program includes pre-submission interactions between developers and FDA as well as additional early opportunities to engage with the agency.[336] The draft guidance provides an overview of all available mechanisms through which an applicant can request feedback or a meeting with FDA regarding potential or planned device applications.[337]
- Refuse to Accept Policy for 510(k)s. The guidance document updates the procedures and criteria FDA intends to use in assessing whether a 510(k) submission meets a minimum threshold of acceptability and therefore should be accepted for substantive review to account for combination drug/device products.[338]
- Acceptance of Clinical Data to Support Medical Device Applications and Submissions. The guidance helps manufacturers understand requirements for data submitted from clinical investigations conducted outside the United States, which must be from investigations conducted in accordance with good clinical practice, including review and approval by an independent ethics committee (IEC) and informed consent from subjects.[339]
- Determination of Substantial Equivalence for 510(k)s. This guidance helps 510(k) submitters demonstrate substantial equivalence and is aimed at improving the predictability, consistency, and transparency of the 510(k) premarket review process, and serves as an aid for evaluating the benefit-risk profile of a new device in comparison to the predicate device.[340]
- 510(k) Third-Party Review Program – draft guidance. This draft guidance describes the factors FDA will use in determining which types of devices are eligible for third-party review, outlines the process for the recognition and suspension of third-party review organizations, and aims to ensure consistent quality of work among approved third-party review organizations.[341]
- Multiple Function Device Products – draft guidance. The draft guidance explains FDA’s proposed regulatory approach and policy for all multiple function device products and clarifies when and how FDA intends to assess the impact of other functions not subject to premarket review.[342]
- Acceptance and Filing Reviews for Premarket Approval Applications. This guidance updates prior PMA filing guidance to clarify requirements for combination drug/device products.[343]
C. Medical Device Safety Action Plan
In April, FDA released the Medical Device Safety Action Plan, outlining its vision for how FDA will encourage innovation that spurs the development of safer devices, promotes earlier detection of risks, and facilitates communication of risk information to physicians and patients.[344] To realize this objective, the plan sets out five areas of focus:
- Establish a robust medical device patient safety net in the United States;
- Explore regulatory options to streamline and modernize timely implementation of post-market mitigations;
- Spur innovation towards safer medical devices;
- Advance medical device cybersecurity; and
- Integrate the CDRH premarket and post-market offices and activities to advance the use of a Total Product Life Cycle (“TPLC”) approach to device safety.[345]
Of particular interest is FDA’s proposal to address the threat of security breaches or other cybersecurity vulnerabilities that could harm patients who are using devices with advanced interconnected technologies, such as implantable cardiac pacemakers. FDA’s plan will consider new requirements for developers, including built-in update mechanisms in medical devices and the development of a “Software Bill of Materials,” which would be used by health care providers and consumers to determine how the device functions, what software is required, and the technology upon which the device relies.[346] FDA also is exploring the creation of a public-private partnership called the CyberMed Safety (Expert) Analysis Board, the primary role of which would be to assess vulnerability, evaluate risks, and investigate suspected or confirmed device compromises at the request of FDA or manufacturers.[347]
The plan emphasizes continuing FDA’s work to establish the National Evaluation System for Health Technology (“NEST”), an active surveillance and evaluation system, developed via a public-private partnership, which would complement existing device vulnerability coordination and response mechanisms.[348] NEST’s capabilities will “help improve the quality of real-world evidence that FDA can use to detect emerging safety signals quickly and take appropriate actions.”[349] FDA also intends to consider whether it has the authority to impose special controls to address new or increased risks of devices, more quickly than through rulemaking.[350]
D. FDA Guidance on Devices with Cybersecurity Risk
In October 2018, FDA published draft guidance containing recommendations to help ensure that device manufacturers are adequately addressing evolving cybersecurity threats. This draft guidance builds on the premarket cybersecurity guidance FDA issued in 2014, and is aimed to ensure that marketed medical devices are sufficiently resilient to evolving cybersecurity threats.[351] The draft guidance provides recommendations regarding device design, labeling, and the documentation that should be included in premarket submissions for devices with cybersecurity risk.[352]
The guidance aims to assist manufacturers to: (1) “employ a risk-based approach to the design and development of medical devices with appropriate cybersecurity protections”; (2) “take a holistic approach to device cybersecurity by assessing risks and mitigations throughout the product’s lifecycle”; (3) “ensure maintenance and continuity of critical device safety and essential performance”; and (4) “promote the development of trustworthy devices to help ensure the continued safety and effectiveness of the devices.”[353]
Also in October 2018, FDA became party to a multi-agency agreement that includes the Department of Homeland Security (“DHS”) implementing a framework for greater coordination and information-sharing about medical device cybersecurity vulnerabilities and threats.[354] FDA also supported the development of a Medical Device Cybersecurity Regional Incident Preparedness and Response Playbook.[355] The playbook provides tools and resources to enable health care delivery organizations to prepare for and respond to medical device cybersecurity incidents, including a standardized approach to response efforts.[356]
E. Drug-Use-Related Software
On November 20, FDA began soliciting public comment on its proposed framework for regulating prescription-drug-use-related software “disseminated by or on behalf of a sponsor that accompanies one or more of the sponsor’s prescription drugs or biologics,” intended to provide clarity to stakeholders and promote innovation.[357] Under this framework, FDA anticipated that in most instances, the prescription-drug-use-related software would not require FDA review before being disseminated. FDA would regulate the output of the software (such as screen displays or audio messages) as promotional drug labeling subject to post-marketing requirements. Prior FDA approval would be required in certain circumstances, such as “when a sponsor seeks to demonstrate that software has an effect on a clinically meaningful outcome and seeks to include information about the software in the FDA-required drug labeling.”[358] The framework would not apply to software independently developed by a third party for use with prescription drugs, unless the drug sponsor licenses the software and disseminates it for use with its drug.[359]
F. Delayed Enforcement Requirements
FDA has issued two guidance documents delaying implementation deadlines, thus giving manufacturers additional time to comply with the requirements at certain UDI and postmarket safety reporting requirements.
On November 5, 2018, FDA informed device manufacturers that it was pushing back enforcement of unique device identification (“UDI”) compliance deadlines for at least two years for some Class I and unclassified medical devices, as well as certain other non-sterile devices requiring direct marking.[360] For Class I and certain unclassified medical devices manufactured and labeled on or after September 24, 2018, UDI labeling, standard date format, and Global Unique Device Identification Database data submission requirements will not be enforced until Sept. 24, 2020. FDA also is delaying the direct mark requirement for these devices, which requires devices to bear a UDI on the device itself if the device is reusable and must be reprocessed between uses, by two years until September 24, 2022. Further, FDA will not enforce UDI direct mark requirements for Class III, life-supporting or life-sustaining, and Class II non-sterile devices that were manufactured and labeled prior to their UDI direct mark compliance deadline and remain in inventory, if the UDI “can be derived from other information directly marked on the device.”
The enforcement delays came as a response to FDA’s experience thus far implementing UDI requirements for Class II, Class III, and implantable, life-supporting, or life-sustaining devices.[361] Because FDA expects a similarly high volume of issues and questions from labelers of Class I and unclassified devices, FDA opted to focus on “addressing existing implementation challenges and optimizing the quality and utility of UDI data for higher-risk devices before focusing on UDI implementation issues for lower-risk devices.”[362]
FDA also chose to delay reporting requirements for Combination Product Applicants, subject to the Combination Product Post-marketing Safety Reporting (“PMSR”) Final Rule.[363] The decision to delay enforcement was intended to ensure that applicants have sufficient time to: (1) update reporting and recordkeeping systems and procedures, including information technology systems; (2) comply with the requirements; and (3) consider the recommendations and technical specifications that FDA intends to provide through guidance to support compliance. In addition to the final guidance delaying the PMSR requirements, FDA also issued draft guidance that provides additional details on PMSR compliance.[364]
G. Device-Related Enforcement Letters
In addition to the enforcement letters related to promotional issues and QSR/cGMP issues noted above, FDA also issued several Warning Letters for other device-related violations, such as violations of submissions, reporting, and postmarket surveillance requirements.[365]
- Becton Dickinson & Company.[366] In January, the manufacturer of blood collection tubes was admonished for numerous alleged violations, including failure to file a 510(k) following significant changes to its products and failure to submit medical device reports to FDA.
- Fujifilm, Olympus Medical Systems Corp., Hoya Corp.[367] In March, FDA issued Warning Letters to the three duodenoscope manufacturers in the United States for failure to comply with requested post-market surveillance studies to assess contamination risk.
- SynCardia Systems LLC.[368] In early April, FDA criticized the manufacturer of a temporary artificial heart system for allegedly violating Medical Device Reporting requirements after failing to report information about a death possibly associated with use of the device.
VIII. CONCLUSION
With drug pricing, patient assistance, and the opioid epidemic in the news frequently, we anticipate that 2019 will see some noteworthy developments for drug and device companies. We will track those events and report back in our next alert. In the meantime, if you have questions regarding the matters addressed above, we would welcome the opportunity to speak with you.
[1] United States’ Mot. to Dismiss Relator’s Second Am. Compl., United States ex rel. Health Choice Grp., LLC v. Bayer Corp., Onyx Pharm., Inc., Amerisourcebergen Corp., & Lash Grp., No. 5:17-CV-126-RWS-CMC (E.D. Tex. Dec. 17, 2018).
[2] Press Release, House Comm. on Oversight & Reform, Oversight Committee Launches Sweeping Drug Price Investigation (Jan. 14, 2019), https://oversight.house.gov/news/press-releases/oversight-committee-launches-sweeping-drug-price-investigation.
[3] U.S. Food & Drug Admin., FDA 2019 Lapse in Funding Information, https://www.fda.gov/AboutFDA/WorkingatFDA/ucm629100.htm (last updated Jan. 18, 2019). (archived here)
[4] See U.S. Dep’t of Justice, Associate Attorney General Rachel Brand, Limiting Use of Agency Guidance Documents In Affirmative Civil Enforcement Cases (Jan. 25, 2018), https://www.justice.gov/file/1028756/download.
[5] See id.
[6] See Memorandum, U.S. Dep’t of Justice, Factors for Evaluating Dismissal Pursuant to 31 U.S.C. 3730(c)(2)(A) (Jan. 10, 2018), https://assets.documentcloud.org/documents/4358602/Memo-for-Evaluating-Dismissal-Pursuant-to-31-U-S.pdf.
[7] U.S. Dep’t of Justice, Justice Manual, Section 4-4.111, U.S. Dep’t of Justice,
https://www.justice.gov/jm/jm-4-4000-commercial-litigation#4-4.111.
[8] See Memorandum, U.S. Dep’t of Justice, Individual Accountability for Corporate Wrongdoing (Sept. 9, 2015), https://www.justice.gov/archives/dag/file/769036/download.
[9] Rod J. Rosenstein, U.S. Deputy Attorney General, Remarks at the American Conference Institute’s 35th International Conference on the Foreign Corrupt Practices Act (Nov. 29, 2018), https://www.justice.gov/opa/speech/deputy-attorney-general-rod-j-rosenstein-delivers-remarks-american-conference-institute-0 (emphasis added).
[10] Certain settlements involving multiple FCA-related theories may appear twice in case totals and recovery amounts.
[11] Publication of OIG Special Advisory Bulletin on Patient Assistance Programs for Medicare Part D Enrollees, 70 Fed. Reg. 70623, 70624 (Nov. 22, 2005), https://oig.hhs.gov/fraud/docs/alertsandbulletins/2005/2005papspecialadvisorybulletin.pdf.
[12] See, e.g., U.S. Dep’t of Health and Human Servs., Office of Inspector Gen., Notice of Modification of OIG Advisory Opinion No. 02-01 at 1 (Mar. 3, 2017), https://oig.hhs.gov/fraud/docs/advisoryopinions/2017/AdvOpn02-1-mod.pdf.
[13] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Drug Maker Actelion Agrees to Pay $360 Million to Resolve False Claims Act Liability for Paying Kickbacks (Dec. 6, 2018), https://www.justice.gov/opa/pr/drug-maker-actelion-agrees-pay-360-million-resolve-false-claims-act-liability-paying.
[14] Id.
[15] Id.
[16] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Drug Maker Pfizer Agrees to Pay $23.85 Million to Resolve False Claims Act Liability for Paying Kickbacks (May 24, 2018), https://www.justice.gov/opa/pr/drug-maker-pfizer-agrees-pay-2385-million-resolve-false-claims-act-liability-paying-kickbacks.
[17] Id.
[18] Id.
[19] Id.
[20] Id.
[21] Id.
[22] Id.
[23] Id.
[24] See Jazz Pharmaceuticals Public Limited Company, Form 10-Q, at 70 (filed May 8, 2018), http://services.corporate-ir.net/SEC.Enhanced/SecCapsule.aspx?c=210227&fid=15625057.
[25] See id.
[26] See Jazz Pharmaceuticals Public Limited Company, Form 10-Q, at 21 (filed Nov. 6, 2018), http://services.corporate-ir.net/SEC.Enhanced/SecCapsule.aspx?c=210227&fid=15903029.
[27] Press Release, Lundbeck, Lundbeck reaches voluntary agreement in principle with U.S. Department of Justice (June 6, 2018), https://investor.lundbeck.com/news-releases/news-release-details/lundbeck-reaches-voluntary-agreement-principle-us-department.
[28] Id.
[29] Corp. Governance, Lundbeck, http://www.lundbeck.com/global/about-us/corporate-governance/risk-management (last modified Feb. 7, 2018).
[30] Press Release, Patient Services Inc., PSI Files Lawsuit Against U.S. Department of Health and Human Services (Jan. 9, 2018), https://www.patientservicesinc.org/news/psi-files-lawsuit-against-u-s-department-of-health-and-human-services-for-violating-its-free-speech-rights.
[31] See Insys Therapeutics, Inc., Form 10-Q, at 17–18 (filed Nov. 9, 2018), https://insysrx.gcs-web.com/static-files/9a1a1a6e-3e61-4ab0-9826-f238f3a0d930; United States’ Compl. in Intervention at 4–5, U.S. ex rel. Maria Guzman v. Insys Therapeutic, Inc., No. 2:13-CV-05861-JLS-AJW (C.D. Cal. Apr. 13, 2018).
[32] United States’ Compl. in Intervention at 4–5, U.S. ex rel. Maria Guzman v. Insys Therapeutic, Inc., No. 2:13-CV-05861-JLS-AJW (C.D. Cal. Apr. 13, 2018).
[33] Id. at 5.
[34] Id. at 13–33.
[35] Press Release, U.S. Dep’t of Justice, U.S. Attorney’s Office, D. of Mass., Former Vice President of Insys Pharmaceuticals Pleads Guilty to Racketeering Scheme (Nov. 28, 2018), https://www.justice.gov/usao-ma/pr/former-vice-president-insys-pharmaceuticals-pleads-guilty-racketeering-scheme.
[36] See Information, United States v. Babich, No. 1:16-cr-10343-ADB, No. 652 (D. Mass. Jan. 3, 2019); Parties’ Agreed Statement of Facts, United States v. Babich, No. 1:16-cr-10343-ADB, ECF No. 668 (D. Mass. Jan. 10, 2019).
[37] Press Release, U.S. Dep’t of Justice, U.S. Attorney’s Office, E. Dist. of Pa., Abbott Laboratories and AbbVie Inc. to Pay $25 Million to Resolve False Claims Act Allegations of Kickbacks and Off-Label Marketing of the Drug TriCor® (Oct. 26, 2018), https://www.justice.gov/usao-edpa/pr/abbott-laboratories-and-abbvie-inc-pay-25-million-resolve-false-claims-act-allegations.
[38] Id.
[39] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Medical Device Maker ev3 to Plead Guilty and Pay $17.9 Million for Distributing Adulterated Device; Covidien Paid $13 Million to Resolve Civil Liability for Second Device (Dec. 4, 2018), https://www.justice.gov/opa/pr/medical-device-maker-ev3-plead-guilty-and-pay-179-million-distributing-adulterated-device.
[40] Press Release, Medtronic, Medtronic Statement Regarding Recent DOJ Announcement (Dec. 4, 2018), https://www.medtronic.com/us-en/about/news/media-resources/medtronic-statement-regarding-doj.html.
[41] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Abiomed, Inc. Agrees to Pay $3.1 Million to Resolve Kickback Allegations (Mar. 8, 2018), https://www.justice.gov/usao-ma/pr/abiomed-inc-agrees-pay-31-million-resolve-kickback-allegations.
[42] Id.
[43] For example, in 2015 Daiichi Sankyo Inc. paid $39 million to resolve allegations that it made improper payments to physicians for “speaking” on duplicative topics at Daiichi-paid dinners and for lavish dinners that exceeded the company’s own internal cost limitations. See Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Daiichi Sankyo Inc. Agrees to Pay $39 Million to Settle Kickback Allegations Under the False Claims Act (Jan. 9, 2015), https://www.justice.gov/opa/pr/daiichi-sankyo-inc-agrees-pay-39-million-settle-kickback-allegations-under-false-claims-act. Forest Laboratories LLC and its subsidiary likewise paid $38 million in December 2016 to resolve allegations of improper kickbacks, including allegations that meals with physicians surpassed company cost limitations. See Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Forest Laboratories and Forest Pharmaceuticals to Pay $38 million to Resolve Kickback Allegations Under the False Claims Act (Dec. 15, 2016), https://www.justice.gov/opa/pr/forest-laboratories-and-forest-pharmaceuticals-pay-38-million-resolve-kickback-allegations.
[44] See Notice of Intervention for Settlement at 1, U.S. ex rel. Gant Van Der Boom v. Precision Medical Products, Inc., No. 2:15-CV-0428 MCE KJN (E.D. Cal. May 18, 2018).
[45] See Second Amended Complaint at 13–18, Precision Medical Products, No. 2:15-CV-0428 MCE KJN (E.D. Cal. Apr. 7, 2017).
[46] Id. at 17–18.
[47] Id. at 21–24.
[48] Id. at 21–23.
[49] Id. at 25–30.
[50] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, United States Settles False Claims Act Allegations Against Trinity Medical Pharmacy And Principals For More Than $2.2 Million (Aug. 10, 2018), https://www.justice.gov/usao-mdfl/pr/united-states-settles-false-claims-act-allegations-against-trinity-medical-pharmacy-and.
[51] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Durable Medical Equipment Provider Lincare Pays $5.25 Million to Resolve False Claims Act Allegations (Aug. 16, 2018), https://www.justice.gov/usao-sdil/pr/durable-medical-equipment-provider-lincare-pays-525-million-resolve-false-claims-act.
[52] Press Release, U.S. Dep’t of Justice, U.S. Attorney’s Office, N. Dist. of Ga., LivaNova agrees to pay $1.87 Million to resolve False Claims Act allegations arising from improper kickback payments (Dec. 4, 2018), https://www.justice.gov/usao-ndga/pr/livanova-agrees-pay-187-million-resolve-false-claims-act-allegations-arising-improper.
[53] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, AmerisourceBergen Corporation Agrees to Pay $625 Million to Resolve Allegations That it Illegally Repackaged Cancer–Supportive Injectable Drugs to Profit From Overfill (Oct. 1, 2018), https://www.justice.gov/opa/pr/amerisourcebergen-corporation-agrees-pay-625-million-resolve-allegations-it-illegally.
[54] Id.
[55] Id.
[56] Id.
[57] Id.
[58] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Alere to Pay U.S. $33.2 Million to Settle False Claims Act Allegations Relating to Unreliable Diagnostic Testing Devices (Mar. 23, 2018), https://www.justice.gov/opa/pr/alere-pay-us-332-million-settle-false-claims-act-allegations-relating-unreliable-diagnostic.
[59] Id.
[60] Id.
[61] Press Release, U.S. Dep’t of Justice, U.S. Attorney’s Office, Dist. of Md., Allergan to Pay $3.5 Million to Settle False Claims Act Allegations Relating to LAP-BAND Bariatric Medical Device (Apr. 16, 2018), https://www.justice.gov/usao-md/pr/allergan-pay-35-million-settle-false-claims-act-allegations-relating-lap-band-bariatric.
[62] Id.
[63] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Medical Device Maker AngioDynamics Agrees to Pay $12.5 Million to Resolve False Claims Act Allegations (July 18, 2018), https://www.justice.gov/opa/pr/medical-device-maker-angiodynamics-agrees-pay-125-million-resolve-false-claims-act.
[64] Id.
[65] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Government Settles $1.2 Million Lawsuit Against Florida Compounding Pharmacy And Its Owner For Excessive Charges To TRICARE (Sept. 28, 2018), https://www.justice.gov/usao-mdfl/pr/government-settles-12-million-lawsuit-against-florida-compounding-pharmacy-and-its.
[66] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Medical Equipment Company Agrees to Pay $5.25 Million to Resolve Allegations of Fraudulent Claims for Compounded Medical Creams (Oct. 22, 2018), https://www.justice.gov/usao-edky/pr/medical-equipment-company-agrees-pay-525-million-resolve-allegations-fraudulent-claims.
[67] Id.
[68] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Attorney General Sessions Announces New Prescription Interdiction & Litigation Task Force (Feb. 27, 2018), https://www.justice.gov/opa/pr/attorney-general-sessions-announces-new-prescription-interdiction-litigation-task-force.
[69] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Deputy Associate Attorney General Stephen Cox Delivers Remarks at the Federal Bar Association Qui Tam Conference (Feb. 28, 2018), https://www.justice.gov/opa/speech/deputy-associate-attorney-general-stephen-cox-delivers-remarks-federal-bar-association.
[70] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Deputy Assistant Attorney General James M. Burnham Delivers Remarks to the 2018 Food and Drug Law Institute Conference (Dec. 13, 2018), https://www.justice.gov/opa/speech/deputy-assistant-attorney-general-james-m-burnham-delivers-remarks-2018-food-and-drug-law.
[71] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Attorney General Jeff Sessions Announces the Formation of Operation Synthetic Opioid Surge (S.O.S.) (July 12, 2018), https://www.justice.gov/opa/pr/attorney-general-jeff-sessions-announces-formation-operation-synthetic-opioid-surge-sos.
[72] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Attorney General Sessions Announces Opioid Fraud and Abuse Detection Unit (Aug. 2, 2017), https://www.justice.gov/opa/pr/attorney-general-sessions-announces-opioid-fraud-and-abuse-detection-unit.
[73] Compl. in Intervention ¶ 4, United States v. Insys Therapeutics, Inc., No. CV 13-5861 JLS (AJWx) (C.D. Cal. Apr. 13, 2018).
[74] See Insys Therapeutics, Inc., Quarterly Report (Form 10-Q), at 17–18 (Nov. 9, 2018); Third Amended Complaint at 7, U.S. ex rel. Maria Guzman v. Insys Therapeutic, Inc., No. 2:13-CV-05861-JLS-AJW (C.D. Cal., Aug. 13, 2018).
[75] Id. at 13–33.
[76] Press Release, U.S. Dep’t of Justice, Former Vice President of Insys Pharmaceuticals Pleads Guilty to Racketeering Scheme (Nov. 28, 2018), https://www.justice.gov/usao-ma/pr/former-vice-president-insys-pharmaceuticals-pleads-guilty-racketeering-scheme.
[77] See Information, United States v. Babich, No. 1:16-cr-10343-ADB (D. Mass, Jan. 3, 2019); Parties’ Agreed Statement of Facts, Babich, No. 1:16-cr-10343-ADB (D. Mass, Jan. 10, 2019).
[78] Mallinckrodt PLC, Annual Report (Form 10-K), at 125 (Feb. 27, 2018); Endo International PLC, Current Report (Form 8-K), at 8.01 (Jan. 11, 2018); Allergan plc, Annual Report (Form 10-K), at F-107 (Feb. 16, 2018).
[79] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Department of Justice Files Motion in Multi-District Opioid Case (Apr. 2, 2018), https://www.justice.gov/opa/pr/department-justice-files-motion-multi-district-opioid-case.
[80] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Justice Department to File Statement of Interest in Opioid Case (Feb. 27, 2018), https://www.justice.gov/opa/pr/justice-department-file-statement-interest-opioid-case.
[81] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Two Chinese Nationals Charged with Operating Global Opioid and Drug Manufacturing Conspiracy Resulting in Deaths (Aug. 22, 2018), https://www.justice.gov/opa/pr/two-chinese-nationals-charged-operating-global-opioid-and-drug-manufacturing-conspiracy.
[82] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Attorney General Sessions Takes Further Action to Combat Opioid Crisis – Directs the DEA to Evaluate Aggregate Production Quotas (Mar. 1, 2018), https://www.justice.gov/opa/pr/attorney-general-sessions-takes-further-action-combat-opioid-crisis-directs-dea-evaluate.
[83] Controlled Substances Quotas, 83 Fed. Reg. 17329 (proposed Apr. 19, 2018) (to be codified at 21 C.F.R. pt. 1303).
[84] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Justice Department, DEA Propose Significant Opioid Manufacturing Reduction in 2019 (Aug. 16, 2018), https://www.justice.gov/usao-edtn/pr/justice-department-dea-propose-significant-opioid-manufacturing-reduction-2019.
[85] Press Release, The White House, President Donald J. Trump Signed H.R. 6 into Law (Oct. 24, 2018), https://www.whitehouse.gov/briefings-statements/president-donald-j-trump-signed-h-r-6-law/.
[86] SUPPORT for Patients and Communities Act, Public Law No: 115-271, H.R. 6, 115th Cong., § 3292 (2017-2018).
[87] Id. § 3273.
[88] Id. § 3012.
[89] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Olympus Medical Systems Corporation, Former Senior Executive Admit Distributing Endoscopes after Failing to File FDA-Required Adverse Event Reports of Serious Infections (Dec. 10, 2018), https://www.justice.gov/usao-nj/pr/olympus-medical-systems-corporation-former-senior-executive-admit-distributing-endoscopes.
[90] Id.
[91] Id.
[92] Id.
[93] Id.
[94] Id.
[95] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Orders Florida Company to Stop Distributing Adulterated and Misbranded Drugs (Mar. 28, 2018), https://www.justice.gov/opa/pr/district-court-orders-florida-company-stop-distributing-adulterated-and-misbranded-drugs.
[96] Id.
[97] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction to Stop New Jersey and New York Companies and Executives From Distributing Unapproved and Misbranded Drugs (Aug. 31, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-stop-new-jersey-and-new-york-companies-and.
[98] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction Against Chicago Companies to Stop Distribution of Adulterated and Misbranded Dietary Supplements and Unapproved and Misbranded Drugs (July 27, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-against-chicago-companies-stop-distribution.
[99] Id.
[100] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Medical Device Maker ev3 to Plead Guilty and Pay $17.9 Million for Distributing Adulterated Device; Covidien Paid $13 Million to Resolve Civil Liability for Second Device (Dec. 4, 2018), https://www.justice.gov/opa/pr/medical-device-maker-ev3-plead-guilty-and-pay-179-million-distributing-adulterated-device.
[101] Id.
[102] Id.
[103] Id.
[104] Id.
[105] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction Against Arkansas Compounding Pharmacy and its CEO to Prevent Distribution of Adulterated Drugs (Apr. 19, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-against-arkansas-compounding-pharmacy-and-its-ceo; Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction Against Mississippi Compounding Pharmacy and Two of its Officers to Prevent Distribution of Adulterated Drugs (June 11, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-against-mississippi-compounding-pharmacy-and-two.
[106] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Orders Tennessee Personal Care Products Manufacturer to Comply With Drug Safety Requirements (Oct. 23, 2018), https://www.justice.gov/opa/pr/district-court-orders-tennessee-personal-care-products-manufacturer-comply-drug-safety.
[107] Id.
[108] Id.
[109] Sandra Moser, Remarks at the American Conference Institute’s 8th Global Forum on Anti-Corruption in High Risk Markets (July 25, 2017).
[110] Press Release, U.S. Sec. and Exch. Comm’n, Sanofi Charged With FCPA Violations (Sept. 4, 2018), https://www.sec.gov/news/press-release/2018-174.
[111] Press Release, U.S. Sec. and Exch. Comm’n, SEC Charges Stryker A Second Time for FCPA Violations (Sept. 28, 2018), https://www.sec.gov/news/press-release/2018-222.
[112] 136 S. Ct. 1989, 2001 (2016) (emphasis added).
[113] United States v. Snap Diagnostics, LLC, No. 1:14-CV-3988, 2018 WL 2689270 (N.D. Ill. June 5, 2018).
[114] Id. at *4.
[115] 840 F.3d 445 (7th Cir. 2016).
[116] 862 F.3d 890 (9th Cir. 2017).
[117] Id. at 906.
[118] Petition for Writ of Certiorari, Gilead, 862 F.3d 890 (No. 17-936).
[119] Id. at *i (No. 17-936) (quoting Escobar, 136 S. Ct. at 2003).
[120] Brief for the United States as Amicus Curiae, Gilead, 862 F.3d 890 (No. 17-936).
[121] Id. at *15.
[122] No. 17-1014, 2018 WL 3949031 (3d Cir. 2018) (unpublished).
[123] Id. at *1.
[124] Id. at *3.
[125] Id. at *4–5.
[126] Id. at *6.
[127] United States ex rel. Solis v. Millennium Pharm., Inc., 885 F.3d 623 (9th Cir. 2018).
[128] Id. at 629.
[129] Id.
[130] Id.
[131] Id.
[132] 898 F.3d 1267, 1273–75 (11th Cir. 2018).
[133] Id. at 1275 (quoting United States ex rel. Clausen v. Lab. Corp. of Am., 290 F.3d 1301, 1311 (11th Cir. 2002)).
[134] Id. at 1277.
[135] Id. at 1277–78.
[136] 31 U.S.C. § 3730(h)(1).
[137] 879 F.3d 71, 73 (3d Cir. 2018).
[138] 129 S. Ct. 2343 (2009).
[139] 133 S. Ct. 2517 (2013).
[140] DiFiore, 879 F.3d at 78.
[141] 31 U.S.C. § 3730(e)(4) (2010).
[142] Id. § 3730(e)(4)(B).
[143] United States ex rel. Forney v. Medtronic, Inc., No. CV 15-6264, 2018 WL 2688424 (E.D. Pa. June 4, 2018).
[144] U.S. ex rel. Moore & Co., P.A. v. Majestic Blue Fisheries, LLC, 812 F.3d 294 (3d Cir. 2016).
[145] Forney, 2018 WL 2688424, at *15 (quoting Moore, 812 F.3d at 306).
[146] United States ex rel. Winkelman v. CVS Caremark Corp., 827 F.3d 201 (1st Cir. 2016).
[147] Forney, 2018 WL 2688424, at *14.
[148] Id. at *15.
[149] Id.
[150] Id. at *16.
[151] Id.
[152] United States ex rel. Wood v. Allergan, Inc., 899 F.3d 163, 165 (2d Cir. 2018) (citing 31 U.S.C. §3730(b)(5)).
[153] Id.
[154] Id. at 167–68.
[155] Id. at 171 (internal citation and quotation marks omitted).
[156] Id. at 175.
[157] Id. at 174.
[158] Id. at 169–70.
[159] Id. at 169.
[160] Scott Gottlieb, U.S. Food & Drug Admin., Statement from FDA Commissioner Scott Gottlieb, M.D., on new efforts to advance medical product communications to support drug competition and value-based health care (June 12, 2018), https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm610415.htm.
[161] Warning Letters 2018: Office of Prescription Drug Promotion, U.S. Food & Drug Admin., https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm594437.htm#OPDP (last viewed Jan. 23, 2019).
[162] See Warning Letters 2017: Office of Prescription Drug Promotion, U.S. Food & Drug Admin., https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm538552.htm#OPDP (last viewed Jan. 23, 2019); see also United States v. Caronia, 703 F.3d 149 (2d Cir. 2012); Amarin Pharma, Inc. v. U.S. Food & Drug Admin., 119 F. Supp. 3d 196 (S.D.N.Y. 2015).
[163] Warning Letters 2018: Office of Prescription Drug Promotion, U.S. Food & Drug Admin., https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm594437.htm#OPDP (last viewed Jan. 23, 2019).
[164] Warning Letter from Koung Lee, RPh, MSHS, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. John F. Weet, PhD, Vice President, Collegium Pharmaceutical, Inc. (Feb. 9, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM597584.pdf.
[165] Id. at 3.
[166] Id. at 1.
[167] Id. at 3.
[168] Warning Letter from Lynn Panholzer, PharmD, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. Peter Honig, MD, MPH, Senior Vice President, Pfizer Inc. (June 19, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM612143.pdf.
[169] Id. at 2, 3.
[170] Id. at 3.
[171] Warning Letter from Rachael Conklin, MS, RN, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. Mamta Puri-Lechner, PhD, Associate Director, Arog Pharmaceuticals, Inc. (June 28, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM612977.pdf.
[172] Id. at 1, 2.
[173] Warning Letter from Lynn Panholzer, PharmD, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. Suzanne Strang, PhD, Executive Director, ASCEND Pharmaceuticals US, LLC (Aug. 16, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM618659.pdf.
[174] Id. at 3.
[175] Letter from Robert Dean, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. Michael Castagna, PharmD, Chief Executive Officer, MannKind Corporation (Oct. 5, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM623557.pdf.
[176] Id. at 3.
[177] Letter from Dhara Shah, PharmD, Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Nada Glavan, Senior Director, Eisai Inc. (Oct. 11, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM623636.pdf.
[178] Id. at 2, 3.
[179] Letter from Andrew S.T. Haffer, Pharm.D., Office of Prescription Drug Promotion, U.S. Food & Drug Admin., to Dr. Polymeropolous, President and Chief Executive Officer, Vanda Pharmaceuticals Inc. (Oct. 22, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM624666.pdf.
[180] Id. at 2, 3.
[181] Warning Letter from Donald J. St. Pierre, Acting Dir., Office of In Vitro Diagnostics & Radiological Health, Ctr. for Devices & Radiological Health, U.S. Food & Drug Admin. to Moshe Becker, Executive Chairman, RADlogics, Inc. (Apr. 5, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm606208.htm.
[182] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Department of Justice Files Complaints Against Florida and California Companies to Stop Use of Experimental Stem Cell Drugs on Patients (May 9, 2018), https://www.justice.gov/opa/pr/department-justice-files-complaints-against-florida-and-california-companies-stop-use.
[183] U.S. Food & Drug Admin., FDA Seeks Permanent Injunctions Against Two Stem Cell Clinics (May 9, 2018), https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607257.htm.
[184] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Department of Justice Files Complaints Against Florida and California Companies to Stop Use of Experimental Stem Cell Drugs on Patients (May 9, 2018), https://www.justice.gov/opa/pr/department-justice-files-complaints-against-florida-and-california-companies-stop-use.
[185] U.S. Food & Drug Admin., Statement from FDA Commissioner Scott Gottlieb, M.D. on the FDA’s New Policy Steps and Enforcement Efforts to Ensure Proper Oversight of Stem Cell Therapies and Regenerative Medicine (Aug. 28, 2017), https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm573443.htm.
[186] Warning Letters 2018: Office of Prescription Drug Promotion, U.S. Food & Drug Admin, https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm594437.htm#OPDP (last viewed July 20, 2018).
[187] U.S. Food & Drug Admin., Statement from FDA Commissioner Scott Gottlieb, M.D., on New Efforts to Advance Medical Product Communications to Support Drug Competition and Value-Based Health Care (June 12, 2018), https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm610415.htm.
[188] Id.
[189] U.S. Food & Drug Admin., Guidance for Industry: Medical Product Communications That Are Consistent With the FDA-Required Labeling – Questions and Answers. (June 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537130.pdf.
[190] Id. at 3.
[191] Id. at 4.
[192] Id. at 5.
[193] Id. at 5, 6.
[194] Id. at 5.
[195] Id. at 8.
[196] Id. at 10.
[197] Id. at 11.
[198] Id. at 12.
[199] Id. at 3.
[200] U.S. Food & Drug Admin., Guidance for Industry: Drug and Device Manufacturer Communications With Payors, Formulary Committees, and Similar Entities – Questions and Answers (June 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537347.pdf.
[201] Id. at 1.
[202] Id. at 4.
[203] Id. at 7.
[204] Id. at 18.
[205] Id. at 18, 19.
[206] Id. at 22.
[207] U.S. Food & Drug Admin., Guidance for Industry: Presenting Quantitative Efficacy and Risk Information in Direct-to-Consumer Promotional Labeling and Advertisements (Oct. 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM623515.pdf.
[208] Id. at 2.
[209] Id. at 4.
[210] Id. at 5.
[211] Id. at 6, 7.
[212] Id. at 7, 8.
[213] Pharmaceutical Information Exchange Act, H.R. 2026, 115th Cong. (2017),
https://www.congress.gov/bill/115th-congress/house-bill/2026.
[214] Id.
[215] Medical Product Communications Act of 2017, H.R. 1703, 115th Cong. (2017),
https://www.congress.gov/bill/115th-congress/house-bill/1703/text.
[216] United States ex rel. Ibanez v. Bristol-Myers Squibb Co., 138 S. Ct. 2582 (Mem) (2018); United States ex rel. King v. Solvay Pharm., Inc., 138 S. Ct. 2030 (Mem) (2018).
[217] United States ex rel. Ibanez, 874 F.3d 905, 915 (6th Cir. 2017); United States ex rel King v. Solvay Pharm., Inc., 871 F.3d 318, 329 (5th Cir. 2017) (per curiam).
[218] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Deputy Assistant Attorney General James M. Burnham Delivers Remarks to the 2018 Food and Drug Law Institute Conference (Dec. 13, 2018), https://www.justice.gov/opa/speech/deputy-assistant-attorney-general-james-m-burnham-delivers-remarks-2018-food-and-drug-law.
[219] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction Against Arkansas Compounding Pharmacy and its CEO to Prevent Distribution of Adulterated Drugs (Apr. 19, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-against-arkansas-compounding-pharmacy-and-its-ceo; see also Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, United States Files Civil Enforcement Action to Stop Arkansas Compounding Pharmacy and CEO from Manufacturing and Distributing Adulterated Drugs (Mar. 1, 2018), https://www.justice.gov/opa/pr/united-states-files-civil-enforcement-action-stop-arkansas-compounding-pharmacy-and-ceo.
[220] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, District Court Enters Permanent Injunction Against Mississippi Compounding Pharmacy and Two of its Officers to Prevent Distribution of Adulterated Drugs (June 11, 2018), https://www.justice.gov/opa/pr/district-court-enters-permanent-injunction-against-mississippi-compounding-pharmacy-and-two.
[221] See U.S. Food & Drug Admin., Warning Letters 2017 (Mar. 14, 2018), https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm538552.htm; see also U.S. Food & Drug Admin., Warning Letters 2018 (Dec. 18, 2018), https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm594437.htm.
[222] Warning Letter from Francis Godwin, Acting Dir., Office of Mfg. Quality, U.S. Food & Drug Admin. to Mr. T.G. Velumani, Chief Executive Officer, Alchymars ICM SM Private Limited (Feb. 16, 2018).
[223] Warning Letter from Francis Godwin, Acting Dir., Office of Mfg. Quality, U.S. Food & Drug Admin. to Mr. Yinghua Liu, President, Lijian Yinghua Biochemical and Pharmaceutical Co., Ltd. (Apr. 19, 2018).
[224] Warning Letter from Francis Godwin, Acting Dir., Office of Mfg. Quality, U.S. Food & Drug Admin. to Ms. Shirley Cai, Chief Executive Officer, Zhuhai United Laboratories Co., Ltd. (June 27, 2018).
[225] Warning Letter from Timothy Stenzel, Dir. Office of In Vitro Diagnostics & Radiological Health, U.S. Food & Drug Admin. to Fredrik O. Dalborg, CEO and Group President, Boule Medical AB (Oct. 2, 2018).
[226] Warning Letter from Karlton T. Watson, Program Division Dir., Office of Biological Prods. Ops. – Div. 2, U.S. Food & Drug Admin. to Rita F. Alexander, Owner/Manager and Jenny R. Galloway, Laboratory & Medical Director, StemGenex Biologic Laboratories, LLC (Oct. 31, 2018).
[227] Warning Letter from Donald St. Pierre, Acting Dir., Office of In Vitro Diagnostics & Radiological Health, Ctr. for Devices & Radiological Health, U.S. Food & Drug Admin. to Raymond Ryu, CEO, Dexcowin Co., Ltd. (Feb. 20, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm599697.htm.
[228] Warning Letter from Shari J. Shambaugh, Program Div. Dir., Div. 3/West, Office of Med. Device & Radiological Health Operations, U.S. Food & Drug Admin. to Galen T. Spooner, Owner, US Vascular, LLC (June 7, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm625034.htm.
[229] Warning Letter from Shari J. Shambaugh, Program Div. Dir., Div. 3/West, Office of Med. Device a& Radiological Health Operations, U.S. Food & Drug Admin. to Ai Chen Teo, President & Gen. Manager, Anigan, Inc. (July 25, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm615066.htm.
[230] Warning Letter from Blake Bevill, Program Div. Dir., Div. 2-Central, Office of Med. Device & Radiological Health Operations, U.S. Food & Drug Admin. to Vincent Forlenza, Chairman & CEO, Becton, Dickinson & Co. (Sept. 14, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm622651.htm.
[231] U.S. Food & Drug Admin., Guidance for Industry: Mixing, Diluting, or Repackaging Biological Products Outside the Scope of an Approved Biologics License Application (Jan. 18, 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM434176.pdf.
[232] Id. at 4.
[233] Id.
[234] Id. at 19–22.
[235] U.S. Food & Drug Admin., Draft Guidance for Industry: Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act (Dec. 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM403496.pdf.
[236] U.S. Good & Drug Admin., Questions and Answers and Guidance for Industry: Data Integrity and Compliance with Drug CGMP (Dec. 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM495891.pdf.
[237] Press Release, U.S. Food & Drug Admin, Statement from FDA Commissioner Scott Gottlieb, M.D., on the agency’s efforts to improve drug quality through vigilant oversight of data integrity and good manufacturing practice (Dec. 12, 2018), https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm628244.htm.
[238] United States ex rel. Greenfield v. Medco Health Sols., Inc., 880 F.3d 89 (3d Cir. 2018).
[239] 42 U.S.C. § 1320a-7b(b)(g).
[240] Greenfield, 880 F.3d at 100.
[241] Report and Recommendation to District Judge, United States ex rel. Medrano et al. v. Diabetic Care RX, LLC, et al., No. 15-cv-62617, ECF No. 100 (S.D. Fla. Nov. 30, 2018).
[242] Id. at 6–7.
[243] Id. at 8–9.
[244] Id. at 20–21.
[245] Id. at 17–18.
[246] 898 F.3d 1267, 1273–75 (11th Cir. 2018).
[247] 42 U.S.C. §1320a-7b(b)(3)(B).
[248] Carrel, 898 F.3d at 1273.
[249] Id. at 1273.
[250] Id. at 1274.
[251] Id.
[252] 318 F. Supp. 3d 1106 (N.D. Ill. 2018).
[253] Id. at 1110.
[254] Id. at 1110–11.
[255] Id. at 1112–13 (emphasis added).
[256] Id. at 1114 (internal quotation and citation marks omitted).
[257] 342 F. Supp. 3d 544, 2018 WL 6567648 (S.D.N.Y Sept. 28, 2018).
[258] Id. at *5.
[259] Id. at *2.
[260] See United States’ Mot. to Dismiss Relator’s Second Am. Compl., United States ex rel. Health Choice Grp., LLC v. Bayer Corp., Onyx Pharm., Inc., Amerisourcebergen Corp., & Lash Grp., No. 5:17-CV-126-RWS-CMC (E.D. Tex. Dec. 17, 2018).
[261] Id. at 3, 14.
[262] Id. at 8, 14.
[263] Id. at 16.
[264] U.S. Dep’t of Health and Human Servs., Office of Inspector Gen., Redacted Final Notice of Rescission 06-04 at 1 (Nov. 28, 2017), https://oig.hhs.gov/fraud/docs/advisoryopinions/2017/AdvOpnRescission06-04.pdf.
[265] Press Release, Office of Pub. Affairs, U.S. Dep’t of Justice, Drug Maker United Therapeutics Agrees to Pay $210 Million to Resolve False Claims Act Liability for Paying Kickbacks (Dec. 20, 2017), https://www.justice.gov/opa/pr/drug-maker-united-therapeutics-agrees-pay-210-million-resolve-false-claims-act-liability.
[266] Complaint, Patient Servs., Inc. v. United States, No. 3:18-cv-00016 (E. D. Va., Jan. 8, 2018).
[267] Id. at 4.
[268] Memorandum In Support Of Defendants’ Mot. For Leave To Take Discovery, Patient Servs., Inc. v. United States, No. 3:18-cv-00016, ECF No. 36 (E.D. Va., July 18, 2018); Order, Patient Servs., Inc. v. United States, No. 3:18-cv-00016, ECF No. 40 (E.D. Va., Jan. 18, 2019).
[269] U.S. Office of Mgmt. & Budget, Exec. Office of the President, RIN No. 0936-AA08, Removal of Safe Harbor Protection for Rebates to Plans or PBMs involving Prescription Pharmaceuticals and Creation of New Safe Harbor Protection (July 18, 2018), https://www.reginfo.gov/public/do/eoDetails?rrid=128288.
[270] Fraud and Abuse; Removal of Safe Harbor Protection for Rebates Involving Prescription Pharmaceuticals and Creation of New Safe Harbor Protection for Certain Point-of-Sale Reductions in Price on Prescription Pharmaceuticals and Certain Pharmacy Benefit Manager Service Fees, 84 Fed. Reg. 2340 (Feb. 6, 2019).
[271] U.S. Dep’t of Health and Human Servs., Trump Administration Proposes to Lower Drug Costs by Targeting Backdoor Rebates and Encouraging Direct Discounts to Patients (Jan. 31, 2019), https://www.hhs.gov/about/news/2019/01/31/trump-administration-proposes-to-lower-drug-costs-by-targeting-backdoor-rebates-and-encouraging-direct-discounts-to-patients.html.
[272] Medicare and State Health Care Programs: Fraud and Abuse; Request for Information Regarding the Anti-Kickback Statute and Beneficiary Inducements CMP, 83 Fed. Reg. 43607 (Aug. 27, 2018).
[273] Id.
[274] Id. at 43609.
[275] Id. at 43609–10.
[276] See 42 C.F.R. § 403.900 et seq.
[277] H.R. 6, 115th Cong., § 6111 (2017-2018).
[278] U.S. Dep’t of Health and Human Servs., Office of Inspector Gen., OIG Advisory Op. 18-02 at 2 (Apr. 30, 2018), https://oig.hhs.gov/fraud/docs/advisoryopinions/2018/AdvOpn18-02.pdf.
[279] Id. at 7.
[280] Id. at 8.
[281] U.S. Dep’t of Health and Human Servs., Office of Inspector Gen., OIG Advisory Op. 18-10 (Sept. 10, 2018), https://oig.hhs.gov/fraud/docs/advisoryopinions/2018/AdvOpn18-10.pdf.
[282] Id. at 2–3.
[283] Id. at 8.
[284] Id. at 8–9.
[285] Id. at 9.
[286] Id. at 9–10.
[287] U.S. Dep’t of Health and Human Servs., Office of Inspector Gen., OIG Advisory Op. 18-14, at 1–2 (Nov. 13, 2018), https://oig.hhs.gov/fraud/docs/advisoryopinions/2018/AdvOpn18-14.pdf.
[288] Id. at 4.
[289] Id.
[290] Id. at 10–12.
[291] U.S. Dept. of Health and Human Servs., American Patients First: The Trump Administration Blueprint to Lower Drug Prices and Reduce Out-of-Pocket Costs (May 11, 2018), https://www.hhs.gov/sites/default/files/AmericanPatientsFirst.pdf.
[292] Press Release, U.S. House of Reps. Committee on Oversight and Reform, Oversight Committee Launches Sweeping Drug Price Investigation (Jan. 14, 2019), https://oversight.house.gov/news/press-releases/oversight-committee-launches-sweeping-drug-price-investigation
[293] Id.
[294] Centers for Medicare & Medicaid Servs.; Proposed Rule, 83 Fed. Reg. 52789 (Oct. 18, 2018).
[295] Id. at 52789–90.
[296] Id. at 52794.
[297] Centers for Medicare & Medicaid Servs.; Advance Notice of Proposed Rulemaking with Comment, 83 Fed. Reg. 54546 (Oct. 30, 2018).
[298] Id. at 54547.
[299] Id.
[300] Centers for Medicare & Medicaid Servs.; Proposed Rule, 83 Fed. Reg. 62152 (Nov. 30, 2018).
[301] Id. at 62152.
[302] Id. at 62158.
[303] Id. at 62153.
[304] Id. at 62168.
[305] Memorandum, U.S. Dept. of Health and Human Servs., Centers for Medicare & Medicaid Servs., Prior Authorization and Step Therapy for Part B Drugs in Medicare Advantage (Aug. 7, 2018), https://www.cms.gov/Medicare/Health-Plans/HealthPlansGenInfo/Downloads/MA_Step_Therapy_HPMS_Memo_8_7_2018.pdf.
[306] Id. at 1.
[307] Id.
[308] Id. at 1–2.
[309] Id. at 3.
[310] Memorandum, U.S. Dept. of Health and Human Servs., Centers for Medicare & Medicaid Servs., Indication-Based Formulary Design Beginning in Contract Year (CY) 2020 (Aug. 29, 2018), https://www.cms.gov/Research-Statistics-Data-and-Systems/Computer-Data-and-Systems/HPMS/Downloads/HPMS-Memos/Weekly/SysHPMS-Memo-2018-Aug-29th.pdf.
[311] Id. at 2.
[312] Id.
[313] Id.
[314] Id. at 2–3.
[315] U.S. Food & Drug Admin., Draft Guidance for Industry: Innovative Approaches for Nonprescription Drug Products (July 2018), https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM613666.pdf.
[316] U.S. Food & Drug Admin., FDA In Brief: FDA issues 63 product-specific guidances to promote generic drug access and drug price competition (Nov. 28, 2018), https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm626952.htm.
[317] U.S. Dep’t of Health & Human Servs., Office of Inspector Gen., HHS Secretary Azar Directs FDA to Establish Working Group on Drug Importation to Address Price Spikes (July 19, 2018), https://www.hhs.gov/about/news/2018/07/19/hhs-secretary-azar-directs-fda-establish-working-group-drug-importation-address-price-spikes.html.
[318] U.S. Food & Drug Admin., Biosimilars Action Plan: Balancing Innovation and Competition (July 2018), https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM613761.pdf.
[319] U.S. Food & Drug Admin., Ctr. for Device & Radiological Health, 2018-2020 Strategic Priorities (Jan. 2018), https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHVisionandMission/UCM592693.pdf.
[320] Jeff Shuren, Charting Our Course for 2018-2020, FDA Voice (Jan. 17, 2018) https://blogs.fda.gov/fdavoice/index.php/2018/01/charting-our-course-for-2018-2020/.
[321] U.S. Food & Drug Admin., Ctr. for Device & Radiological Health, 2018-2020 Strategic Priorities 7 (Jan. 2018), https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHVisionandMission/UCM592693.pdf.
[322] Id.
[323] Id. at 12.
[324] Id. at 17.
[325] Id. at 18.
[326] U.S. Food & Drug Admin. Commissioner Scott Gottlieb, Advancing Policies to Promote Safe, Effective MedTech Innovation, FDA Voice (Dec. 11, 2017), https://blogs.fda.gov/fdavoice/index.php/2017/12/advancing-policies-to-promote-safe-effective-medtech-innovation/.
[327] Statement from FDA Commissioner Scott Gottlieb, M.D. and Jeff Shuren, M.D., Director of the Center for Devices and Radiological Health, on transformative new steps to modernize FDA’s 510(k) program to advance the review of the safety and effectiveness of medical devices (Nov. 26, 2018), https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm626572.htm.
[328] Statement from FDA Commissioner Scott Gottlieb, M.D. and Jeff Shuren, M.D., Director of the Center for Devices and Radiological Health, on transformative new steps to modernize FDA’s 510(k) program to advance the review of the safety and effectiveness of medical devices (Nov. 26, 2018), https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm626572.htm.
[329] Safety and Performance Based Pathway; Guidance for Industry and Food and Drug Administration (Feb. 1, 2019).
[330] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff: The Special 510(k) Program (Sept. 28, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm621682.pdf.
[331] U.S. Food & Drug Admin., FDA In Brief: FDA proposes improvements to the De Novo pathway for novel medical devices to advance safe, effective, and innovative treatments for patients (Dec. 4, 2018), https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm627522.htm; U.S. Food & Drug Admin., Medical Device De Novo Classification Process, 83 Fed. Reg. 63127 (Dec. 7, 2018).
[332] Id.
[333] 83 Fed. Reg. 63127, 63128.
[334] Id.
[335] Id.
[336] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff: Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program (June 7, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm609753.pdf.
[337] Id. at 1.
[338] U.S. Food & Drug Admin., Guidance for Industry and Food and Drug Administration Staff: Refuse to Accept Policy for 510(k)s (Jan. 30, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm315014.pdf.
[339] U.S. Food & Drug Admin., Guidance for Industry and Food and Drug Administration Staff: Acceptance of Clinical Data to Support Medical Device Applications and Submissions (Feb. 21, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm597273.pdf.
[340] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff: Benefit
-Risk Factors to Consider When Determining Substantial Equivalence in Premarket Notifications (510(k)) With
Different Technological Characteristics (Sept. 25, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm404773.pdf.
[341] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff, and Third Party Review Organizations: 510(k) Third Party Review Program (Sept. 14, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm339697.pdf.
[342] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff: Multiple Function Device Products: Policy and Considerations (Apr. 27, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm605683.pdf.
[343] U.S. Food & Drug Admin., Guidance for Industry and Food and Drug Administration Staff: Acceptance and Filing Reviews for Premarket Approval Applications (PMAs) (Jan. 30, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm313368.pdf.
[344] U.S. Food & Drug Admin., Medical Device Safety Action Plan: Protecting Patients, Promoting Public Health (Apr. 17, 2018), https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHReports/UCM604690.pdf.
[345] Id.at 2.
[346] Id. at 13.
[347] Id.
[348] Id. at 6.
[349] Id. at 10.
[350] Id. at 11.
[351] U.S. Food & Drug Admin., FDA In Brief: FDA proposes updated cybersecurity recommendations to help ensure device manufacturers are adequately addressing evolving cybersecurity threats (Oct. 17, 2018), https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm623624.htm
[352] U.S. Food & Drug Admin., Draft Guidance for Industry and Food & Drug Administration Staff: Content of Premarket Submissions for Management of Cybersecurity in Medical Devices (Oct. 18, 2018), https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM623529.pdf.
[353] Id. at 9.
[354] U.S. Food & Drug Admin., DHS-FDA Medical Device Cybersecurity Collaboration: Memorandum of Agreement between the Department of Homeland Security, National Protection and Programs Directorate and the Department of Health and Human Services, Food and Drug Administration, Relating to Medical Device Cybersecurity Collaboration (last updated Oct. 17, 2018), https://www.fda.gov/AboutFDA/PartnershipsCollaborations/MemorandaofUnderstandingMOUs/DomesticMOUs/ucm623568.htm.
[355] The MITRE Corp., Medical Device Cybersecurity: Regional Incident Preparedness and Response Playbook (Oct. 2018), https://www.mitre.org/sites/default/files/publications/pr-18-1550-Medical-Device-Cybersecurity-Playbook.pdf.
[356] Id.
[357] U.S. Food & Drug Admin., FDA In Brief: FDA takes steps to advance a new framework to promote development of digital tools that can inform the safe and effective use of prescription drugs (Nov. 19, 2018), https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm626166.htm?utm_campaign=Federal+Register+Notice+on+Prescription+Drug+Use&utm_medium=email&utm_source=Eloqua.
[358] Id.
[359] U.S. Food & Drug Admin., Prescription Drug-Use-Related Software; Establishment of a Public Docket; Request for Comments, 83 Fed. Reg. 58574 (Nov. 20, 2018), https://www.federalregister.gov/documents/2018/11/20/2018-25206/prescription-drug-use-related-software-establishment-of-a-public-docket-request-for-comments.
[360] U.S. Food & Drug Admin., Immediately in Effect Guidance for Industry and FDA Staff: Unique Device Identification: Policy Regarding Compliance Dates for Class I and Unclassified Devices and Certain Devices Requiring Direct Marking (Nov. 5, 2018), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm592340.pdf
[361] Id. at 4.
[362] Id.
[363] U.S. Food & Drug Admin., Immediately in Effect Guidance for Industry and Food and Drug Administration Staff: Compliance Policy for Combination Product Postmarketing Safety Reporting (Mar. 21, 2018), https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM601461.pdf.
[364] U.S. Food & Drug Admin., Draft Guidance for Industry and Food and Drug Administration Staff: Postmarketing Safety Reporting for Combination Products (Mar. 2018), https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM601454.pdf.
[365] FDA issued 25 device-related Warning Letters, nine of which were written by CDRH. This marks a decrease in activity compared to 2017, when nearly the same number of Warning Letters were issued to device manufacturers in just the second half of the year. Unlike in prior periods, foreign manufacturers marketing their products in the United States were not a common focus; only five such entities received a Warning Letter.
[366] Warning Letter from Joseph Matrisciano, Jr., District Dir., Div. 1/East, N.E. District, Office of Med. Device & Radiological Health Operations, U.S. Food & Drug Admin. to Elizabeth Gaipa, VP Quality Management, Becton Dickinson & Co. (Jan. 11, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm592062.htm.
[367] Warning Letter from William H. Maisel, Acting Dir., Office of Compliance, Ctr. for Devices & Radiological Health, U.S. Food & Drug Admin. to Takaaki Ueda, President & CEO, Fujifilm Med. Systems U.S.A., Inc. (Mar. 9, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm600340.htm; Warning Letter from William H. Maisel, Acting Dir., Office of Compliance, Ctr. for Devices & Radiological Health, U.S. Food & Drug Admin. to Nacho Abia, President & CEO, Olympus Corp. of the Americas (Mar. 9, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm600330.htm; Warning Letter from William H. Maisel, Acting Dir., Office of Compliance, Ctr. for Devices & Radiological Health, U.S. Food & Drug Admin. to David G. Woods, President & CEO, Pentax of America, Inc. (Mar. 9, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm600342.htm.
[368] Warning Letter from Shari J. Shambaugh, Program Div. Dir., Div. 3/West, Office of Med. Device & Radiological Health Operations, U.S. Food & Drug Admin. to Michael P. Garippa, President & CEO, SynCardia Systems, LLC (Apr. 3, 2018), https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm604278.htm.
The following Gibson Dunn lawyers assisted in the preparation of this client update: Stephen Payne, Marian Lee, John Partridge, Jonathan Phillips, Sean Twomey, Reid Rector, Sarah Erickson-Muschko, Allison Chapin, Yamini Grema, Claudia Kraft, Eva Michaels, Stevie Pearl, Julie Hamilton, Jacob Rierson, Emily Riff, Peter Baumann, and Craig Streit.
Gibson Dunn’s lawyers are available to assist in addressing any questions you may have regarding these developments. Please contact the Gibson Dunn lawyer with whom you usually work or any of the following:
Washington, D.C.
Stephen C. Payne, Chair, FDA and Health Care Practice Group (+1 202-887-3693, [email protected])
F. Joseph Warin (+1 202-887-3609, [email protected])
Marian J. Lee (+1 202-887-3732, [email protected])
Daniel P. Chung (+1 202-887-3729, [email protected])
Jonathan M. Phillips (+1 202-887-3546, [email protected])
Los Angeles
Debra Wong Yang (+1 213-229-7472, [email protected])
San Francisco
Charles J. Stevens (+1 415-393-8391, [email protected])
Winston Y. Chan (+1 415-393-8362, [email protected])
New York
Alexander H. Southwell (+1 212-351-3981, [email protected])
Denver
Robert C. Blume (+1 303-298-5758, [email protected])
John D. W. Partridge (+1 303-298-5931, [email protected])
© 2019 Gibson, Dunn & Crutcher LLP
Attorney Advertising: The enclosed materials have been prepared for general informational purposes only and are not intended as legal advice.